drGT:AI 不仅预测药效,还能解释为什么
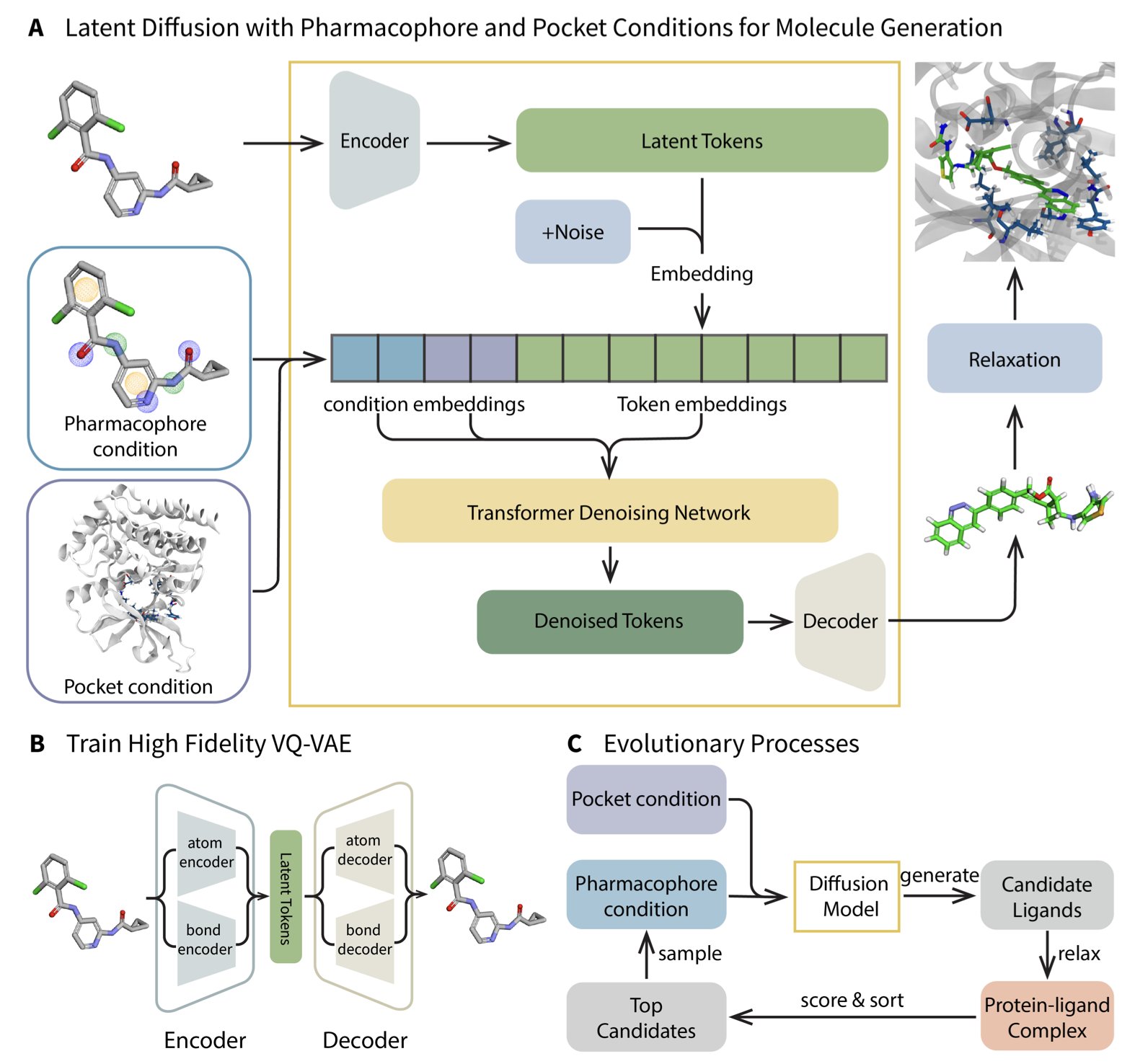
AI 药物研发,PROTAC
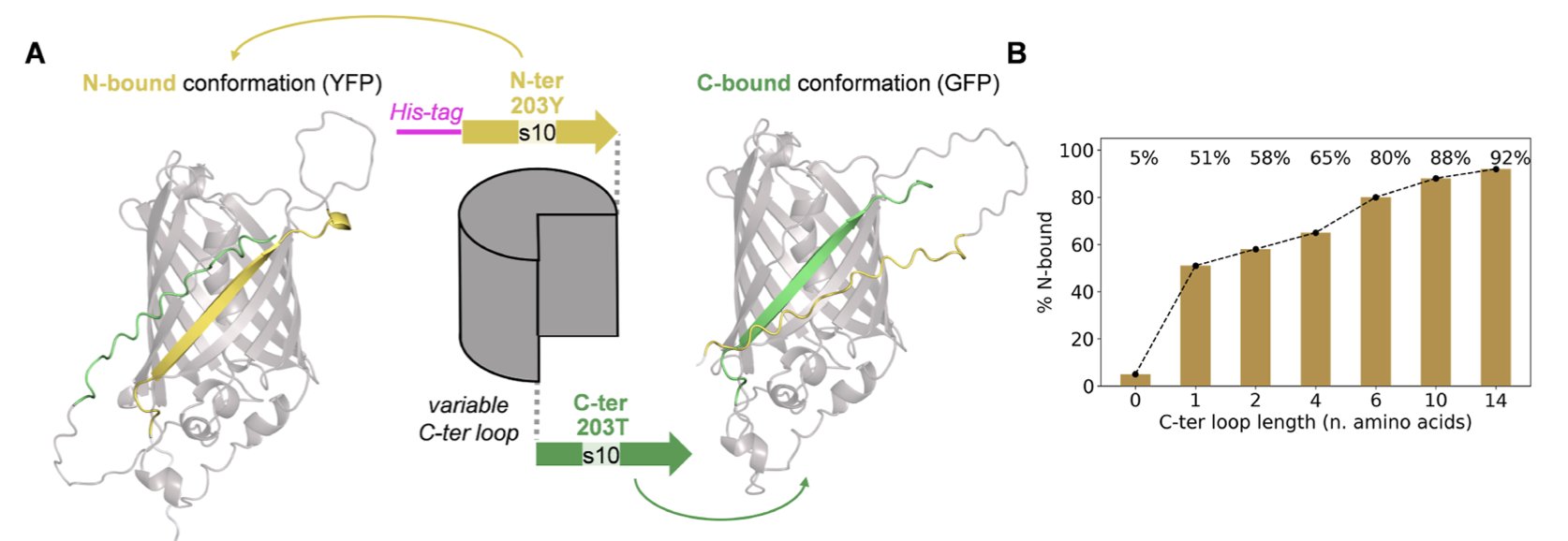
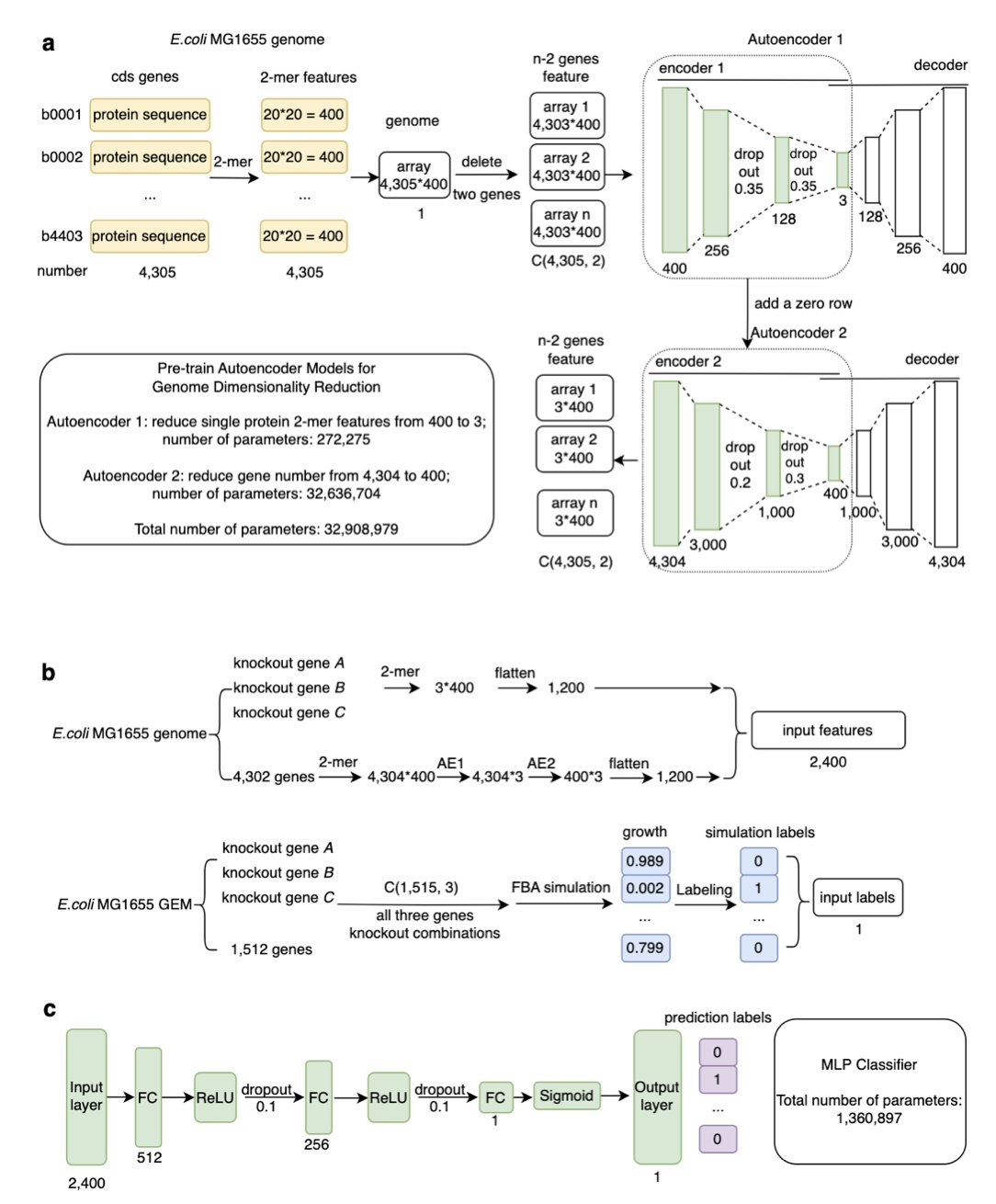
蛋白质语言模型
图注意力网络
可解释性 AI
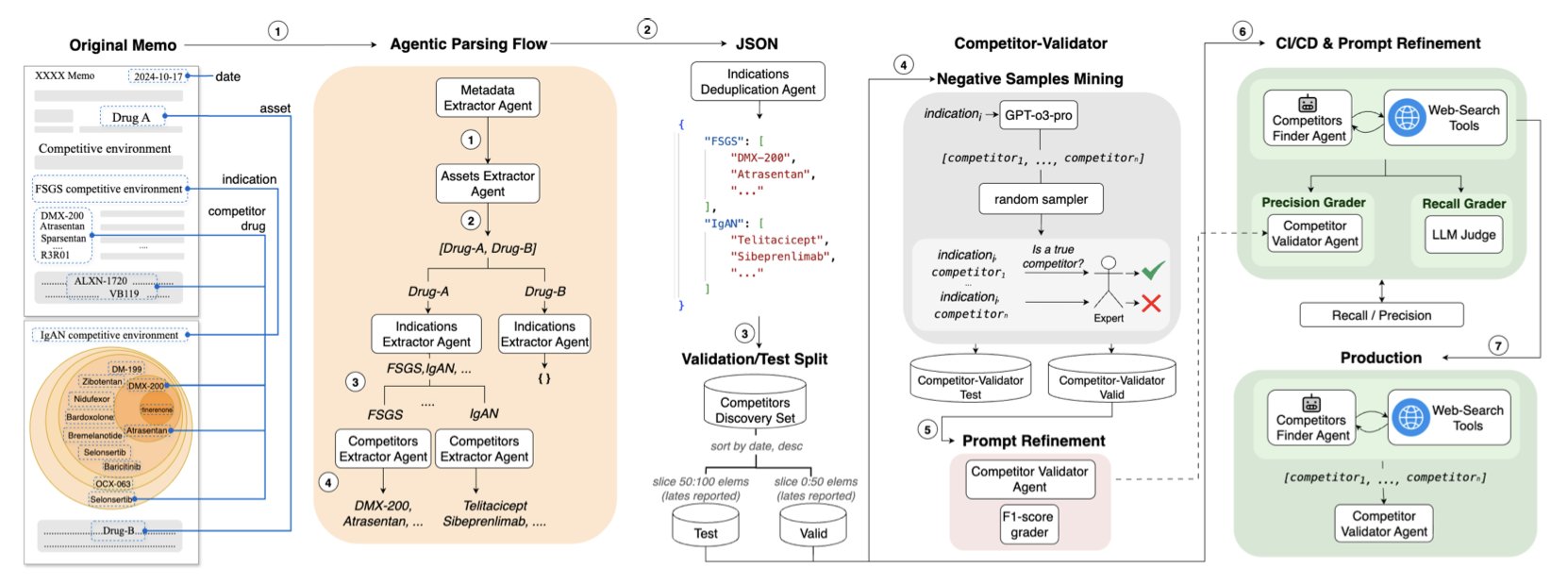
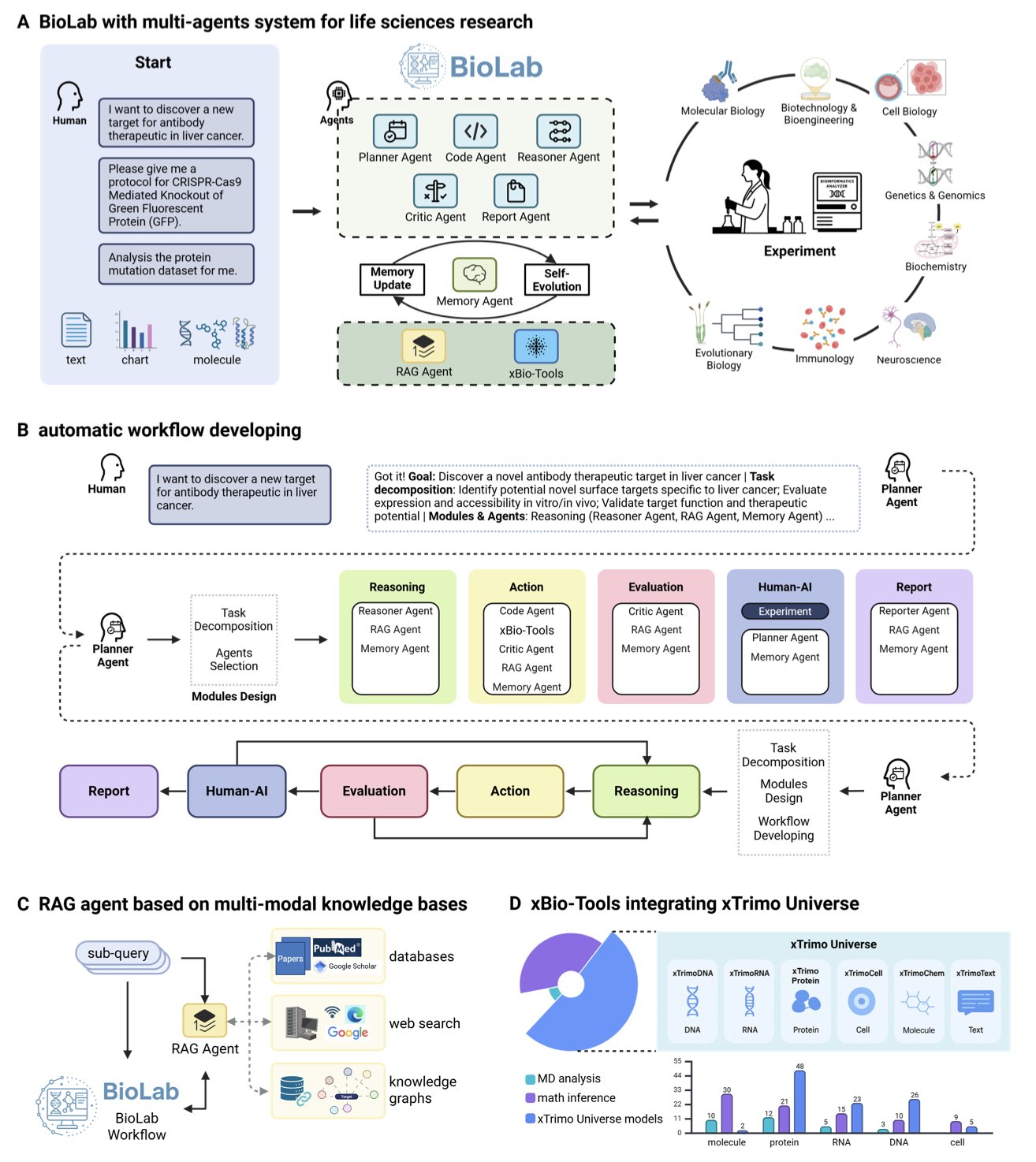
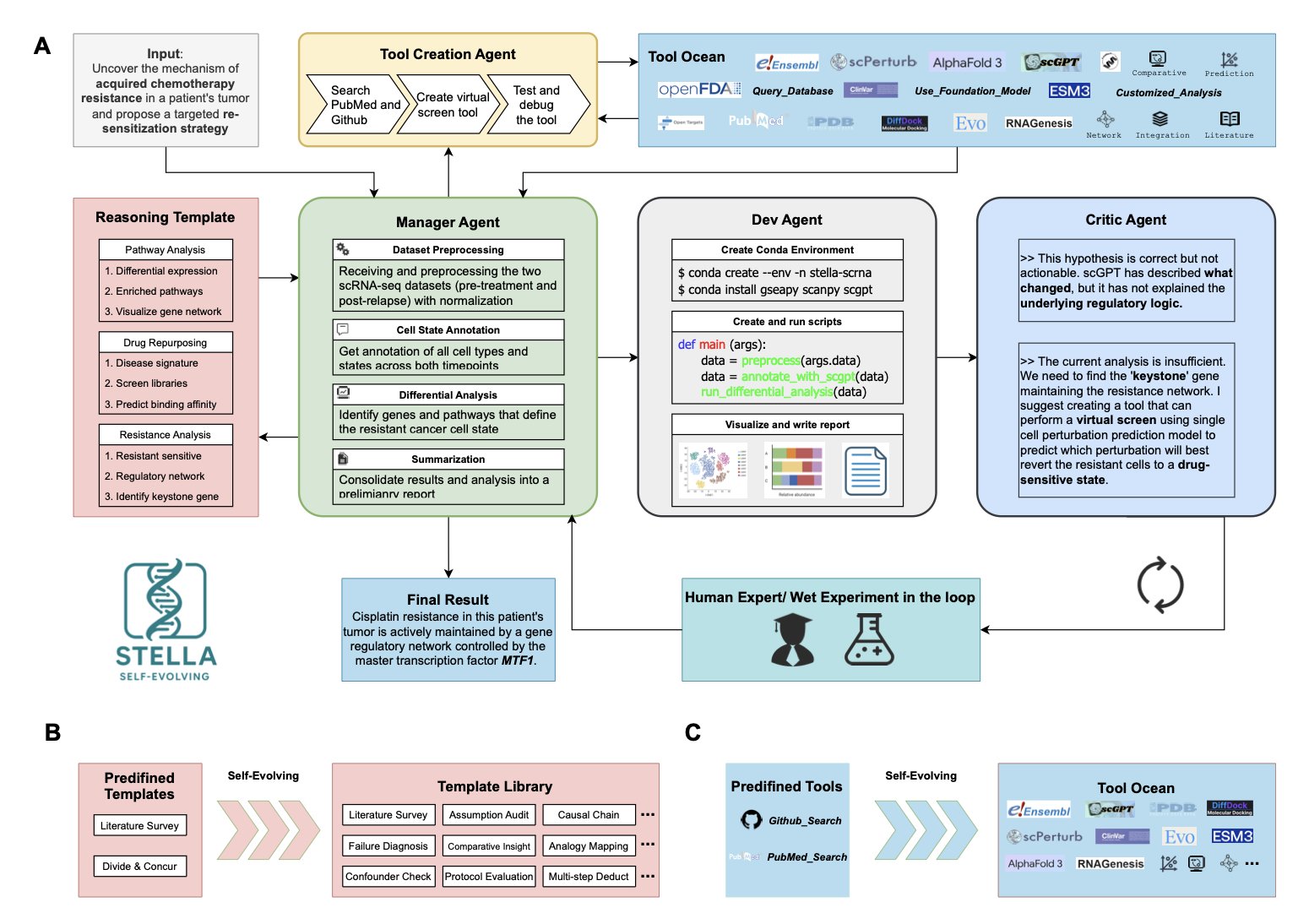
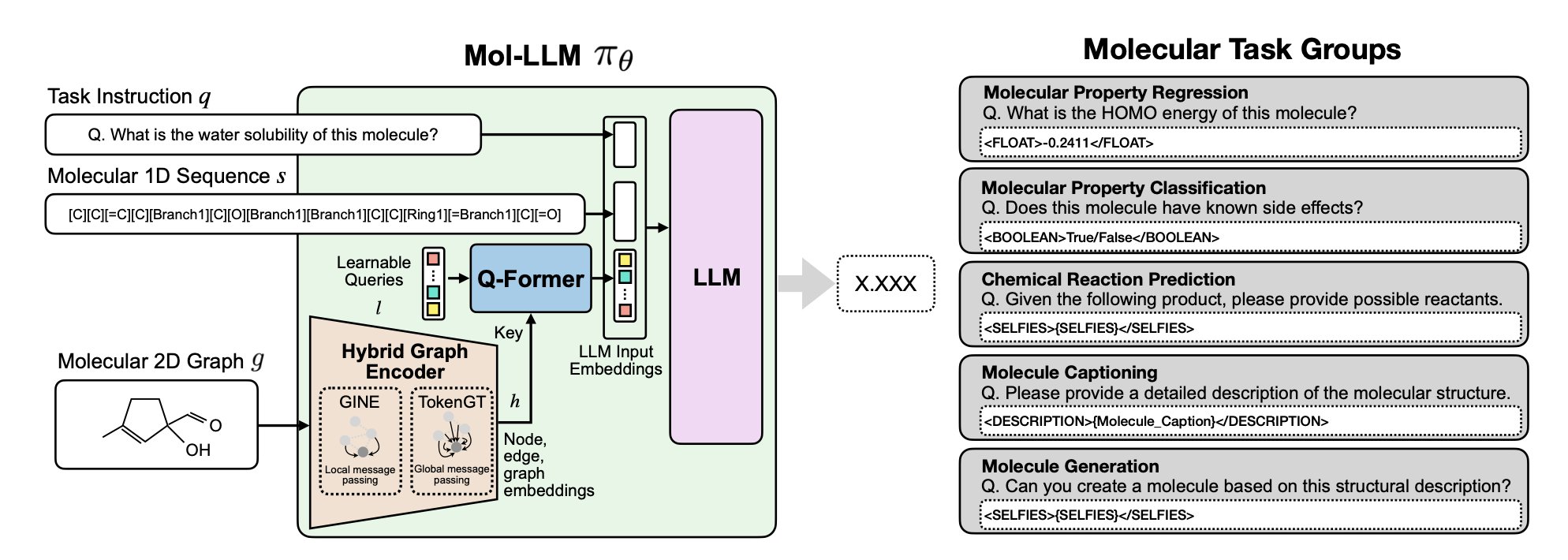
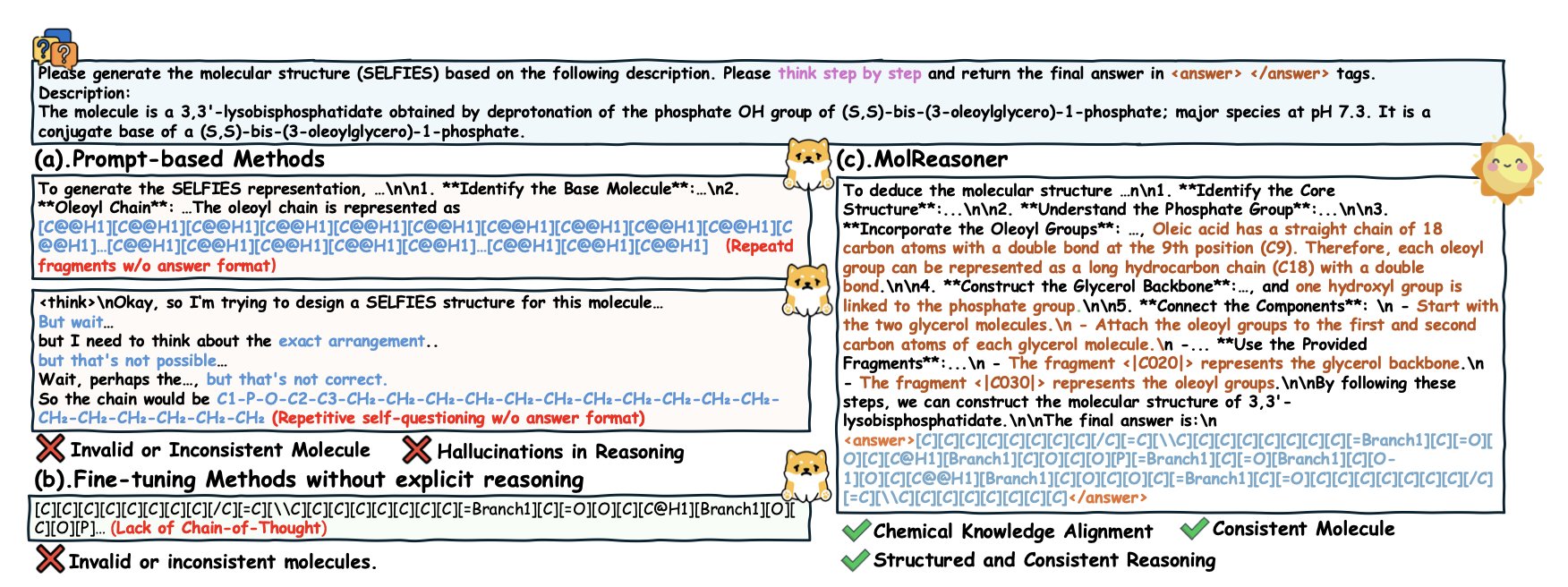
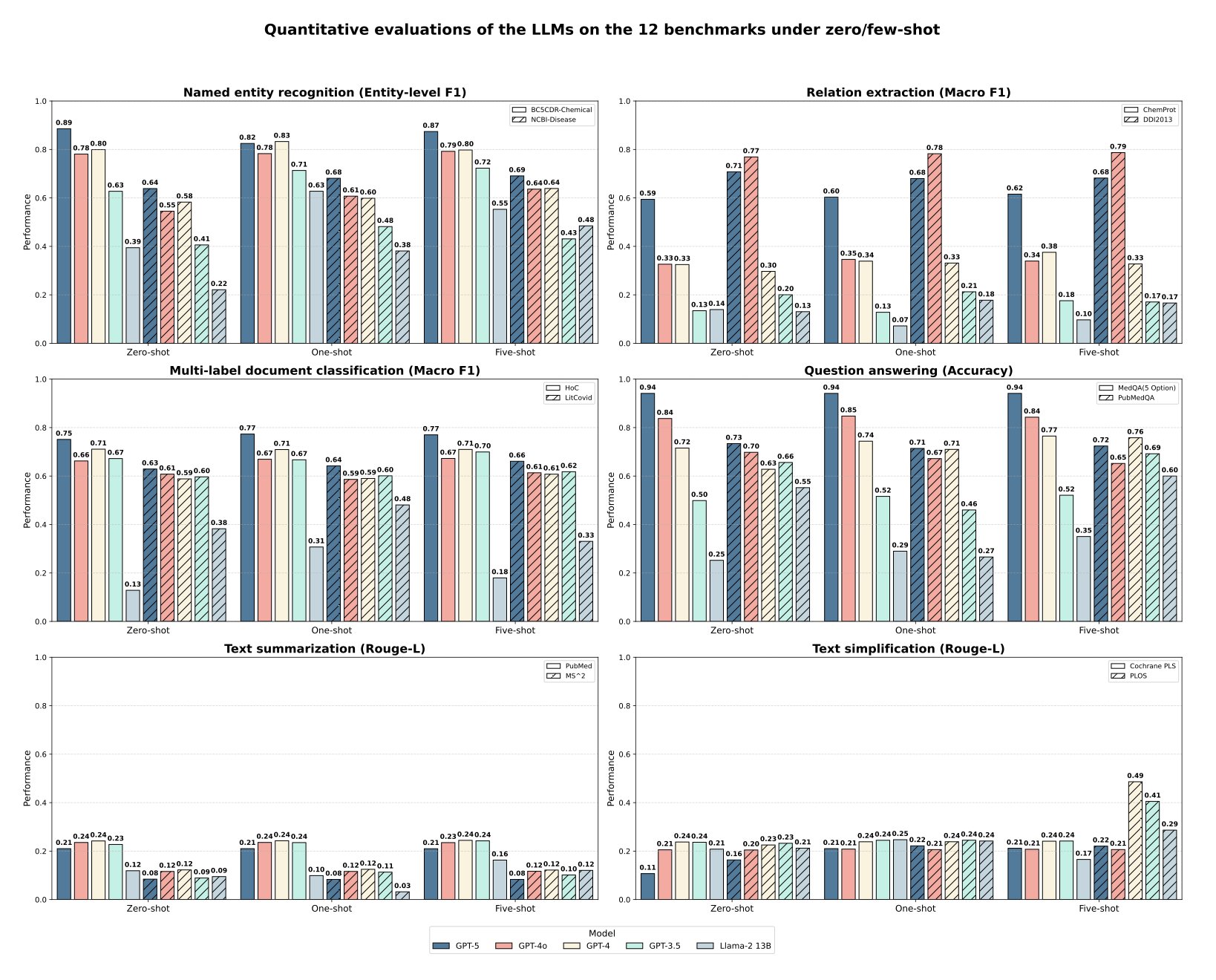
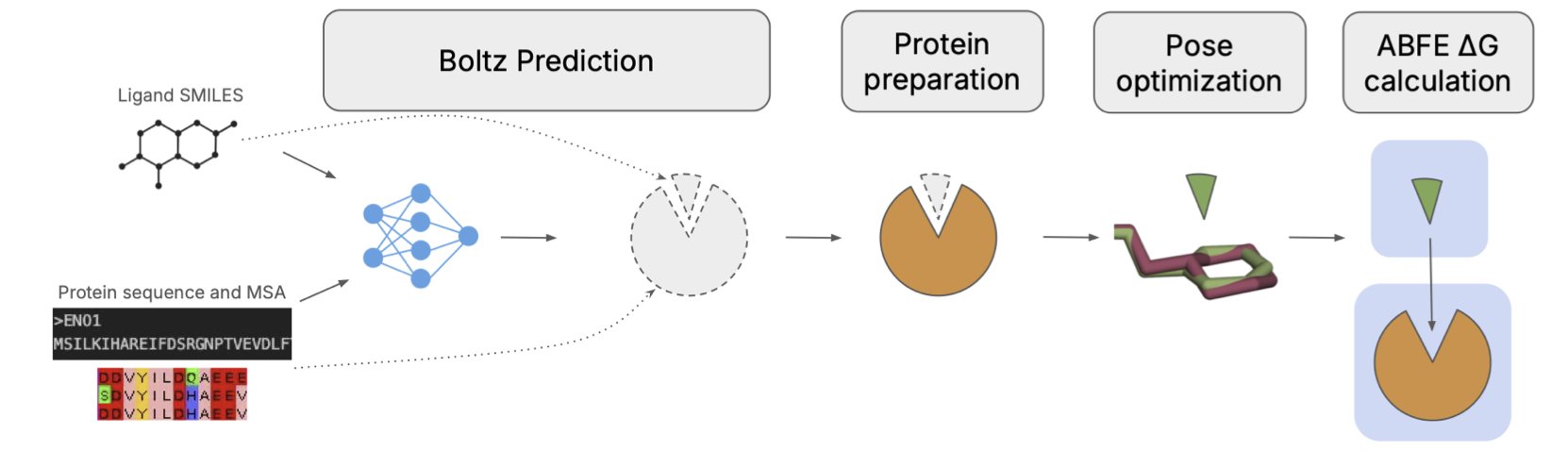
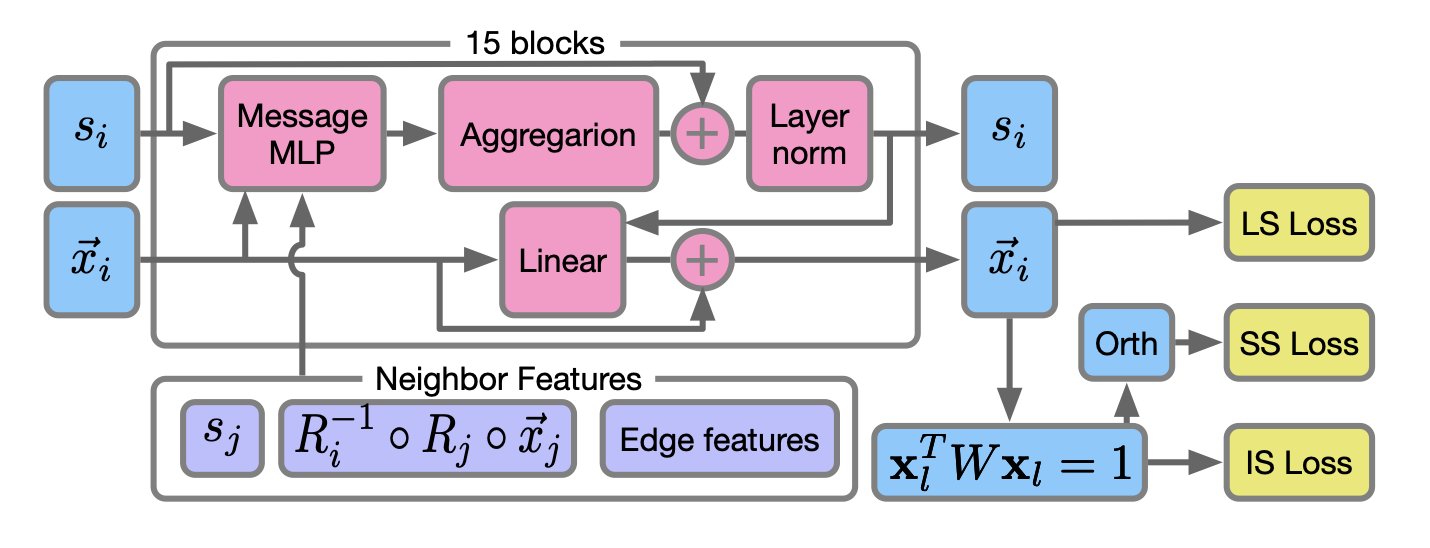
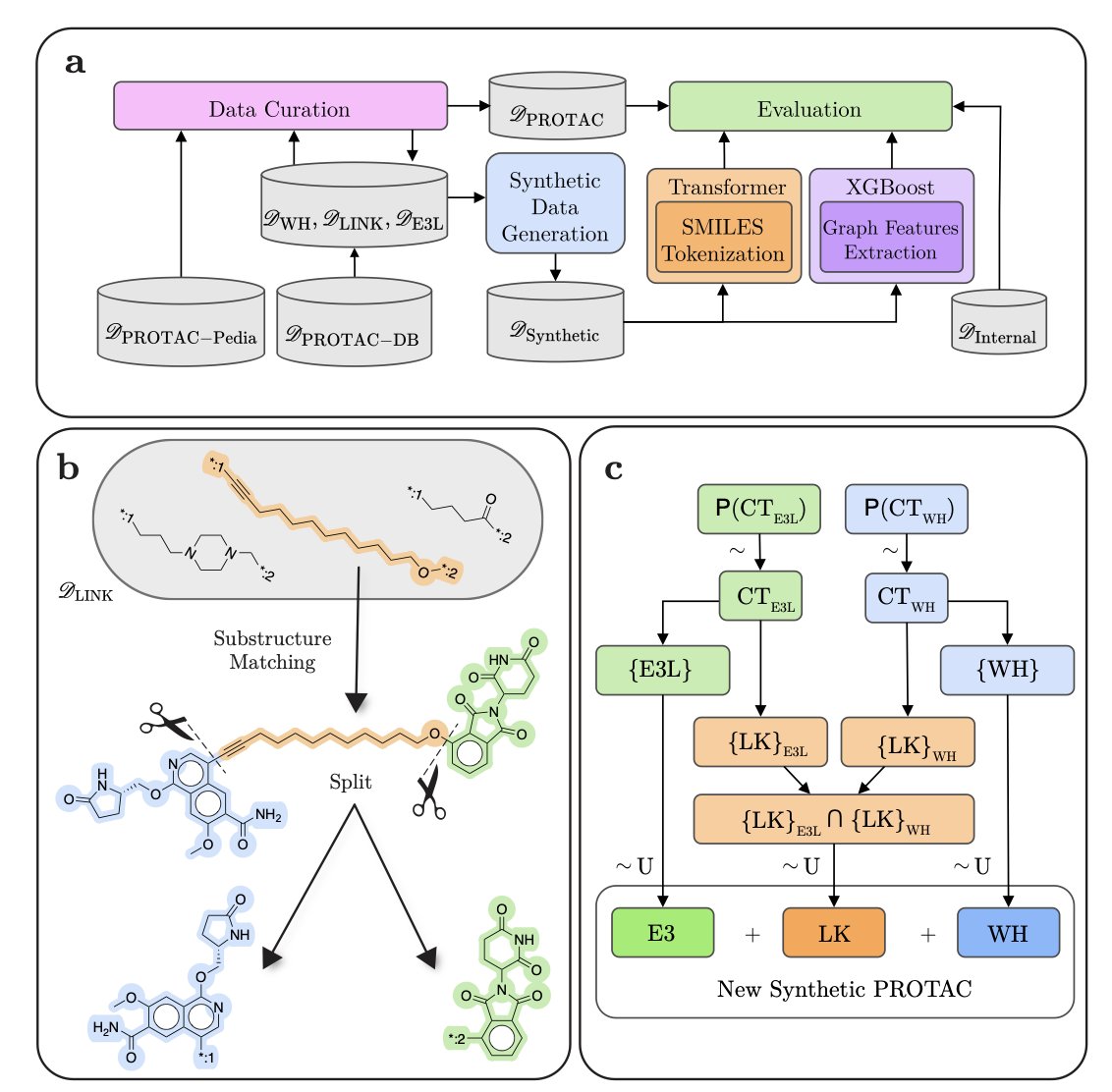
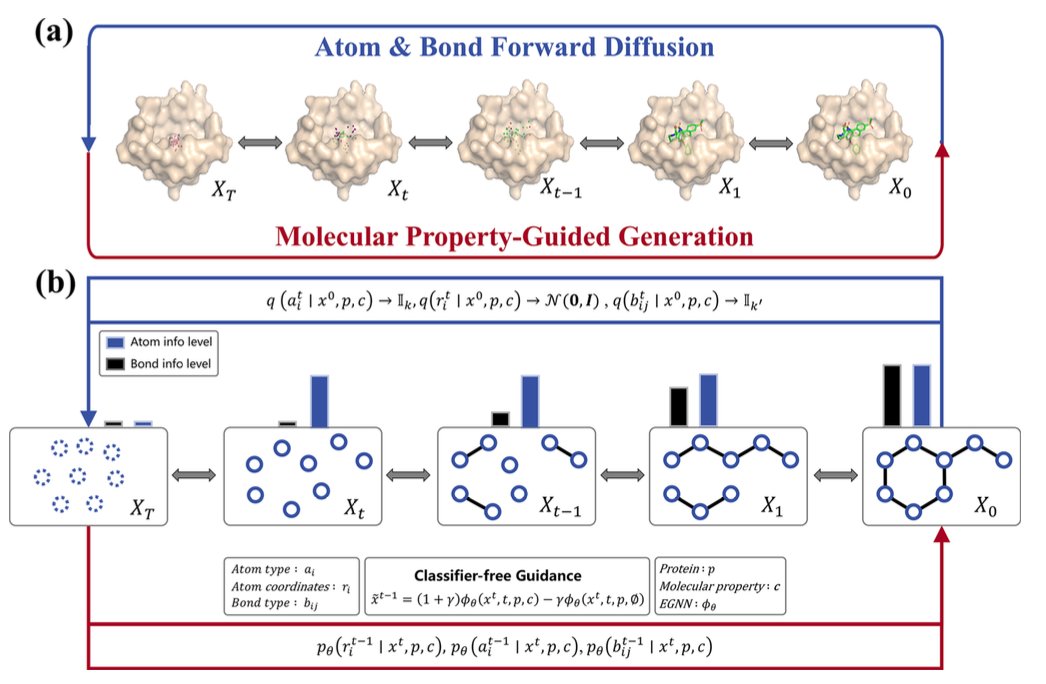
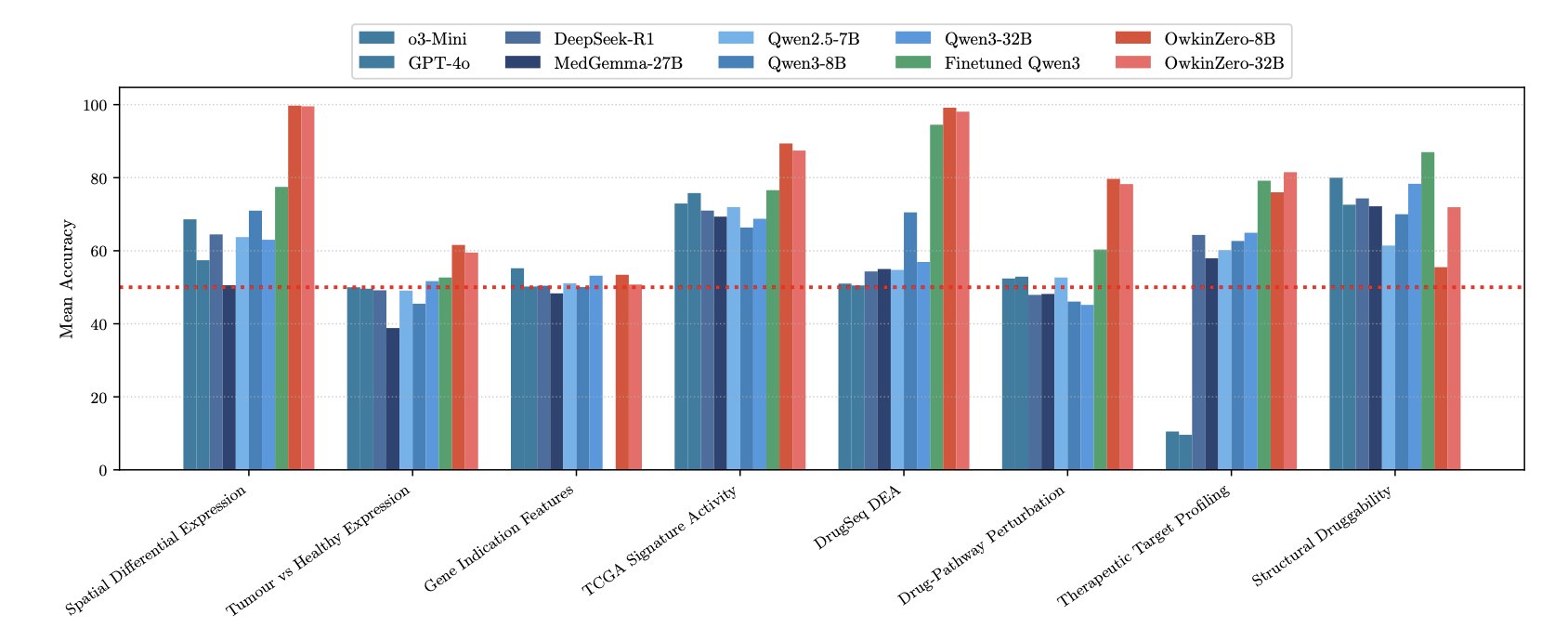
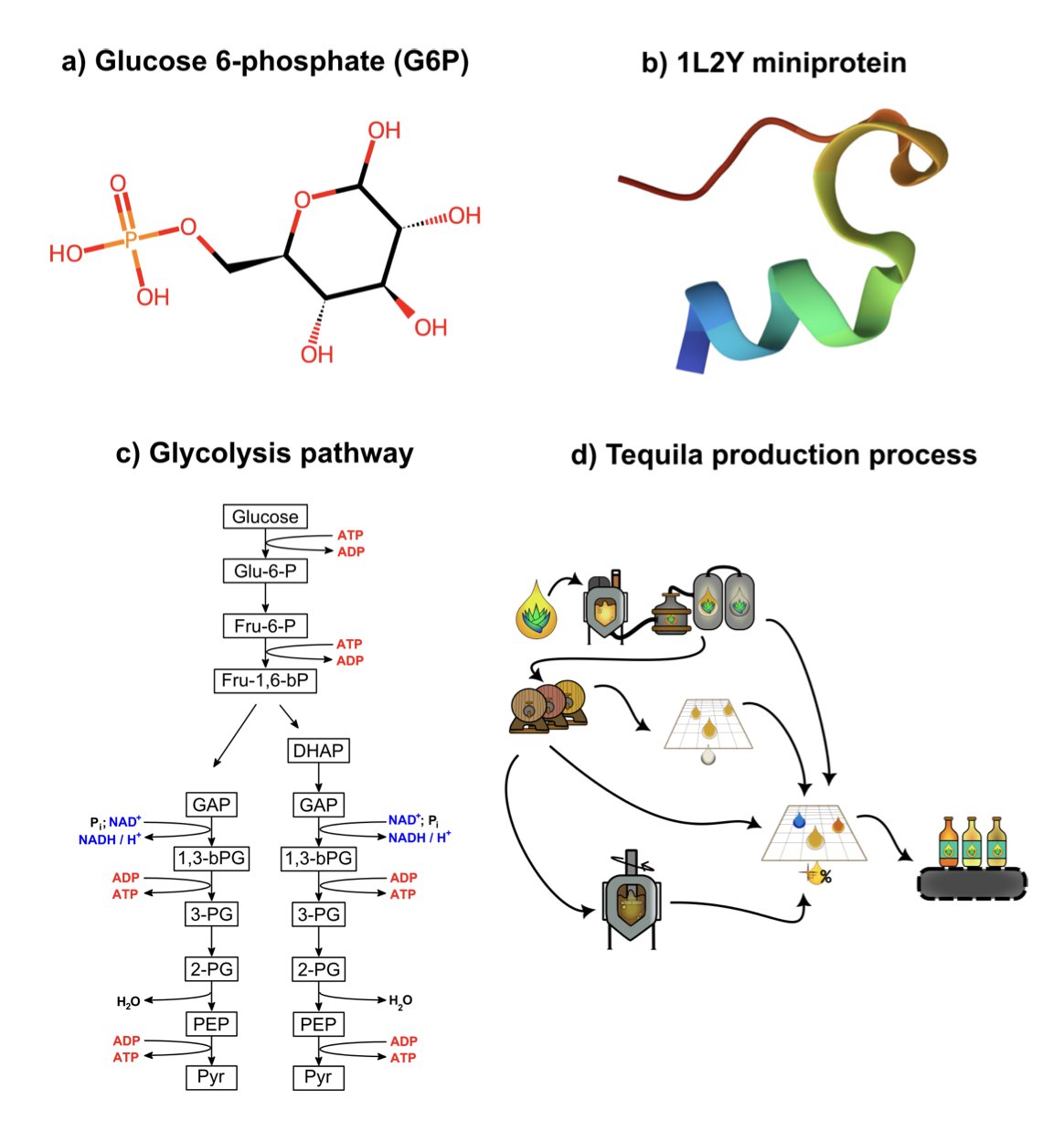
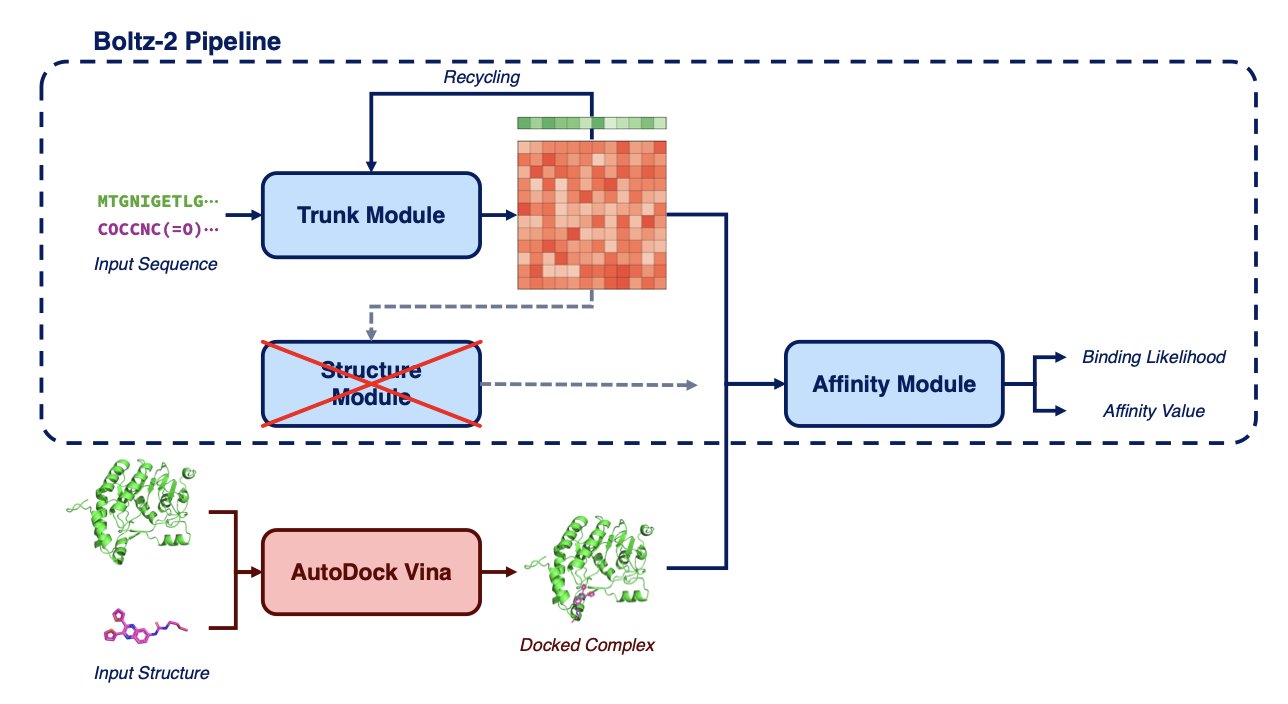
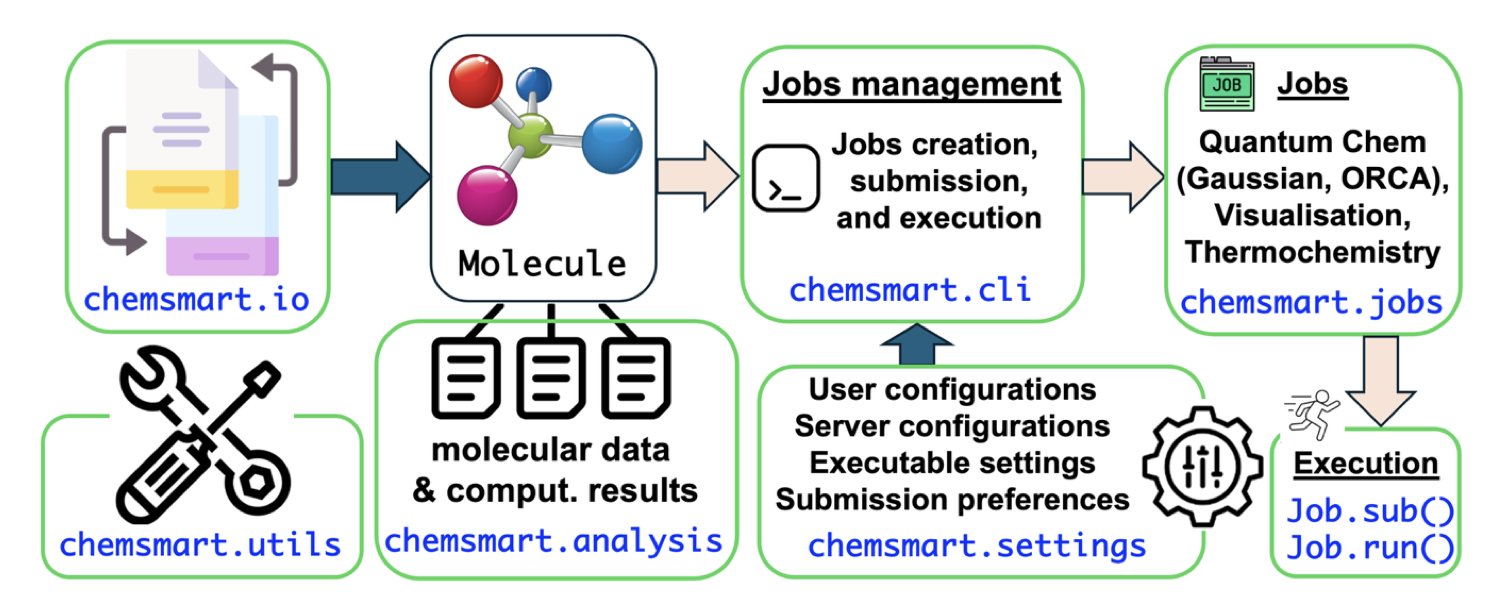
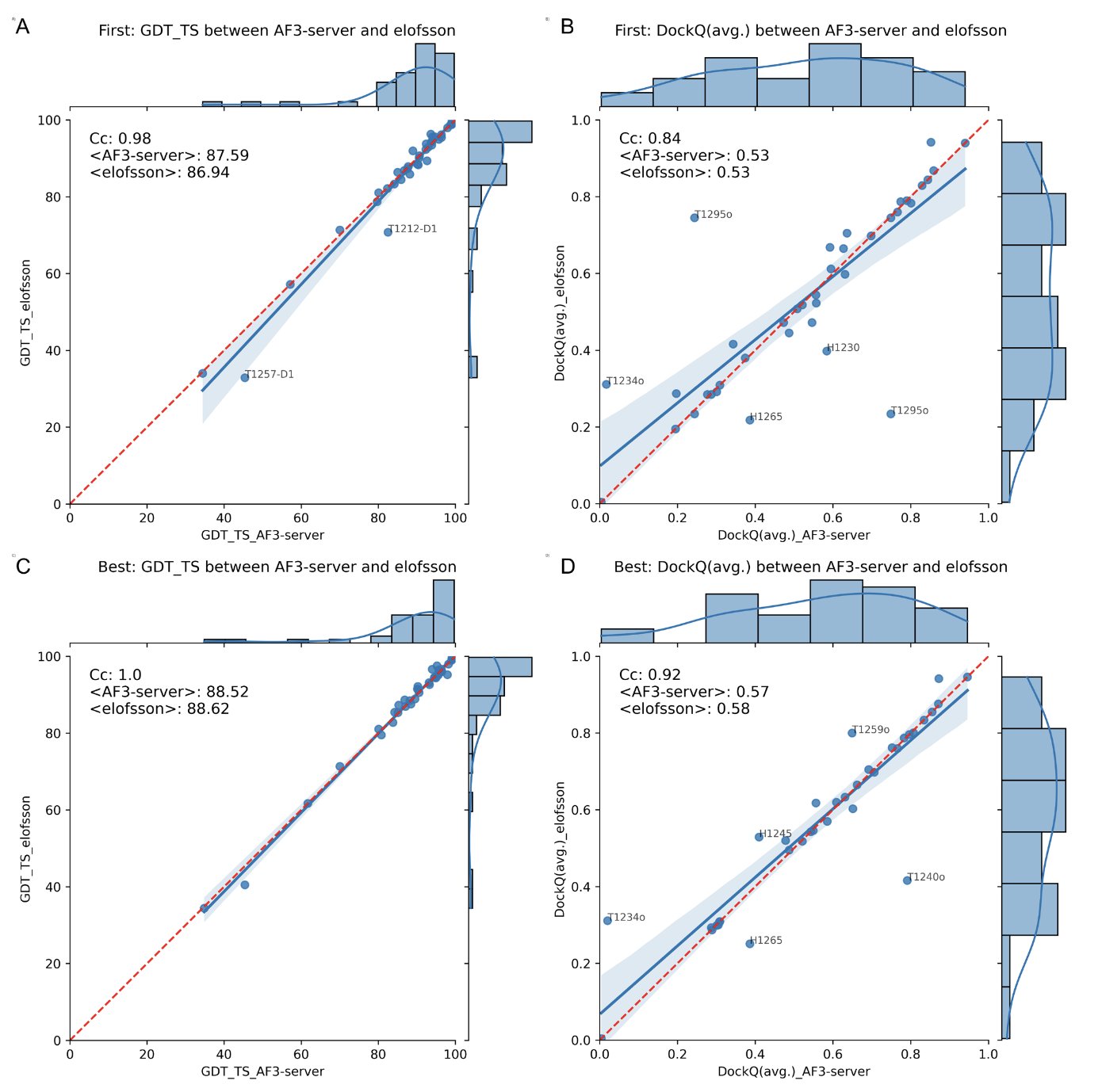
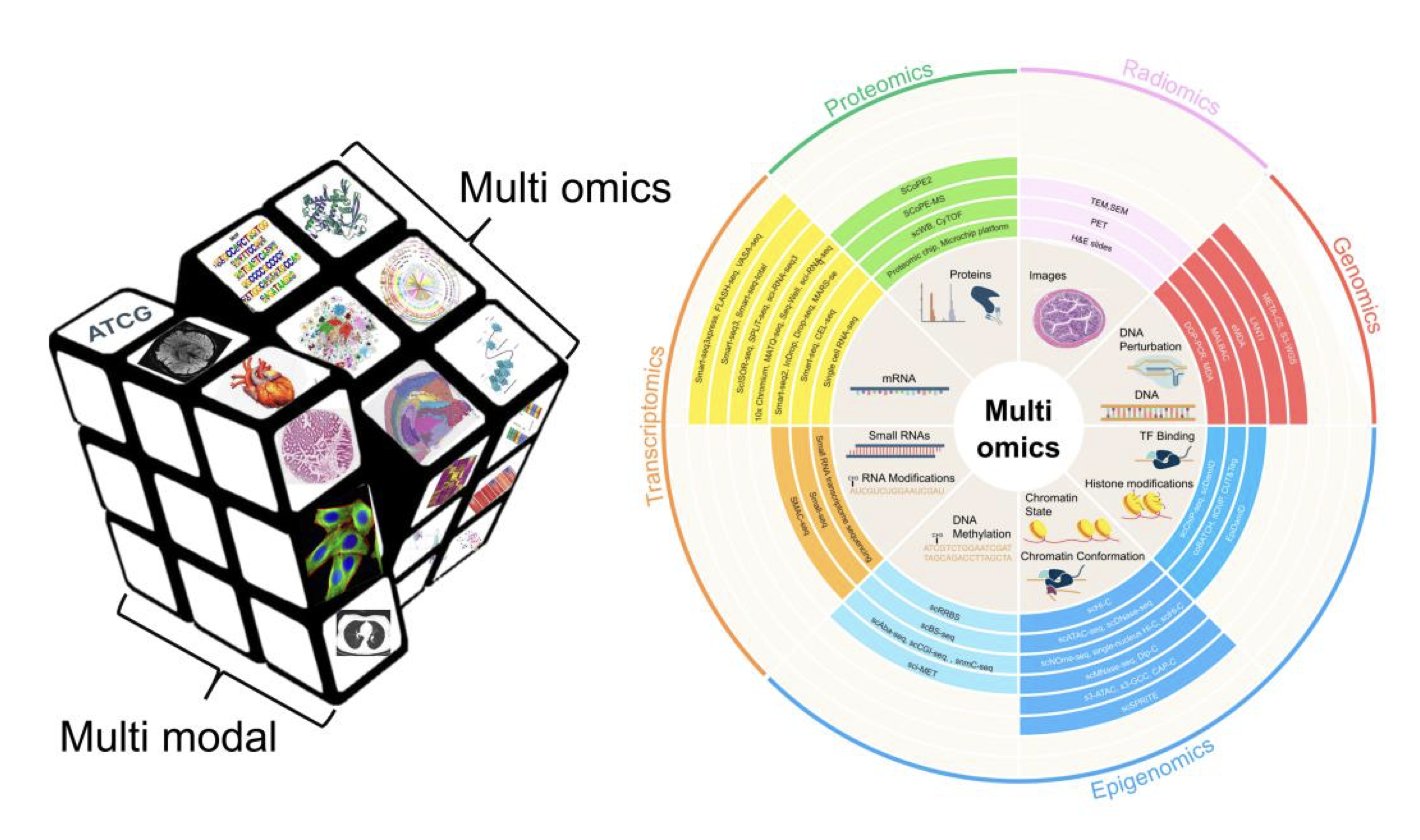
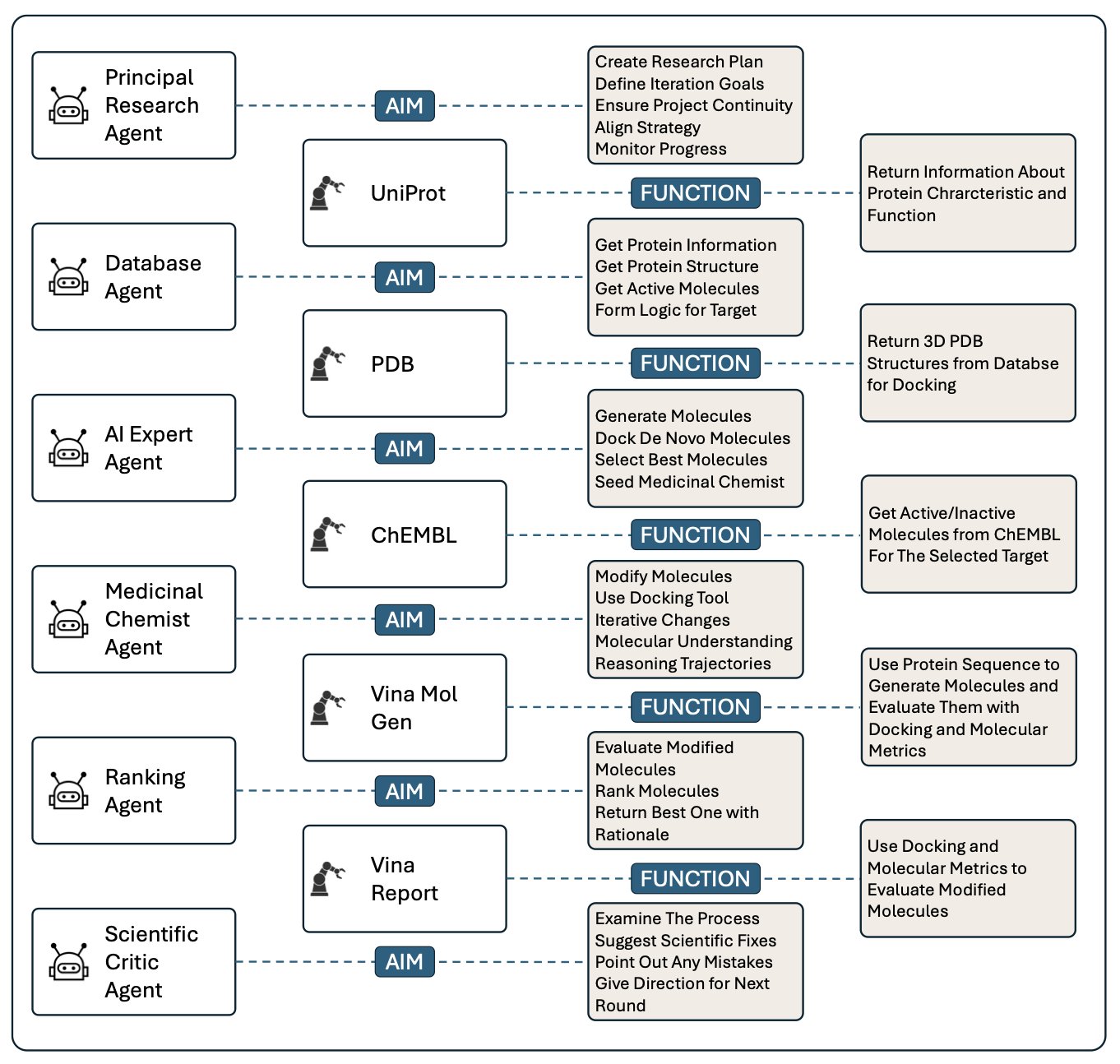
AI 虚拟人:新药研发的终极飞行模拟器
AI 制药,药物研发,机器学习,计算化学,"大语言模型"
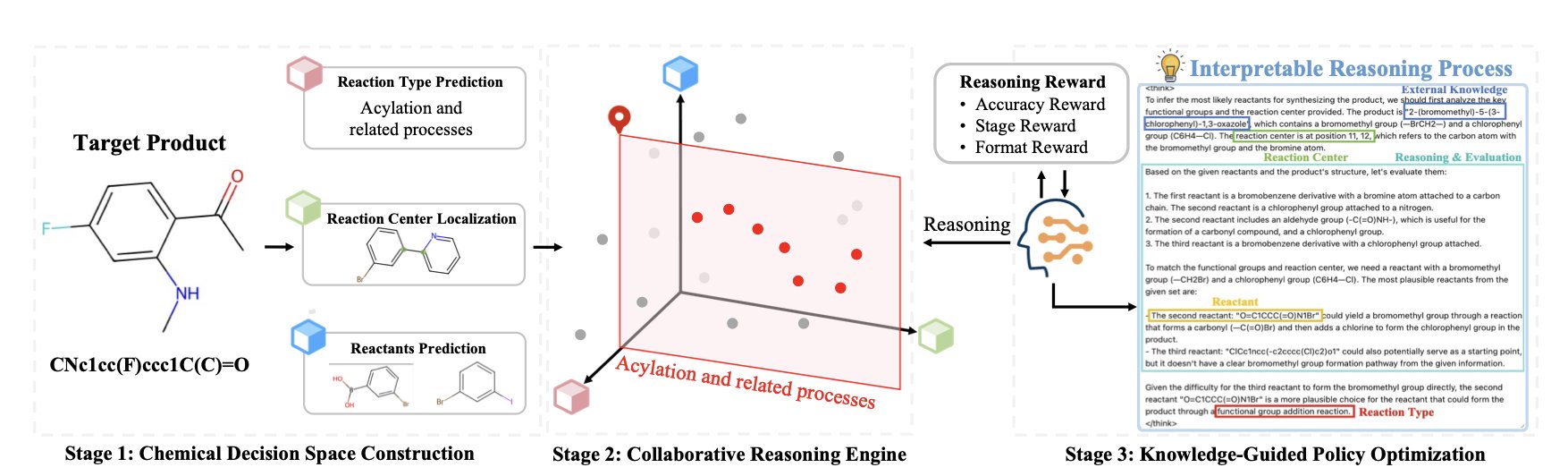
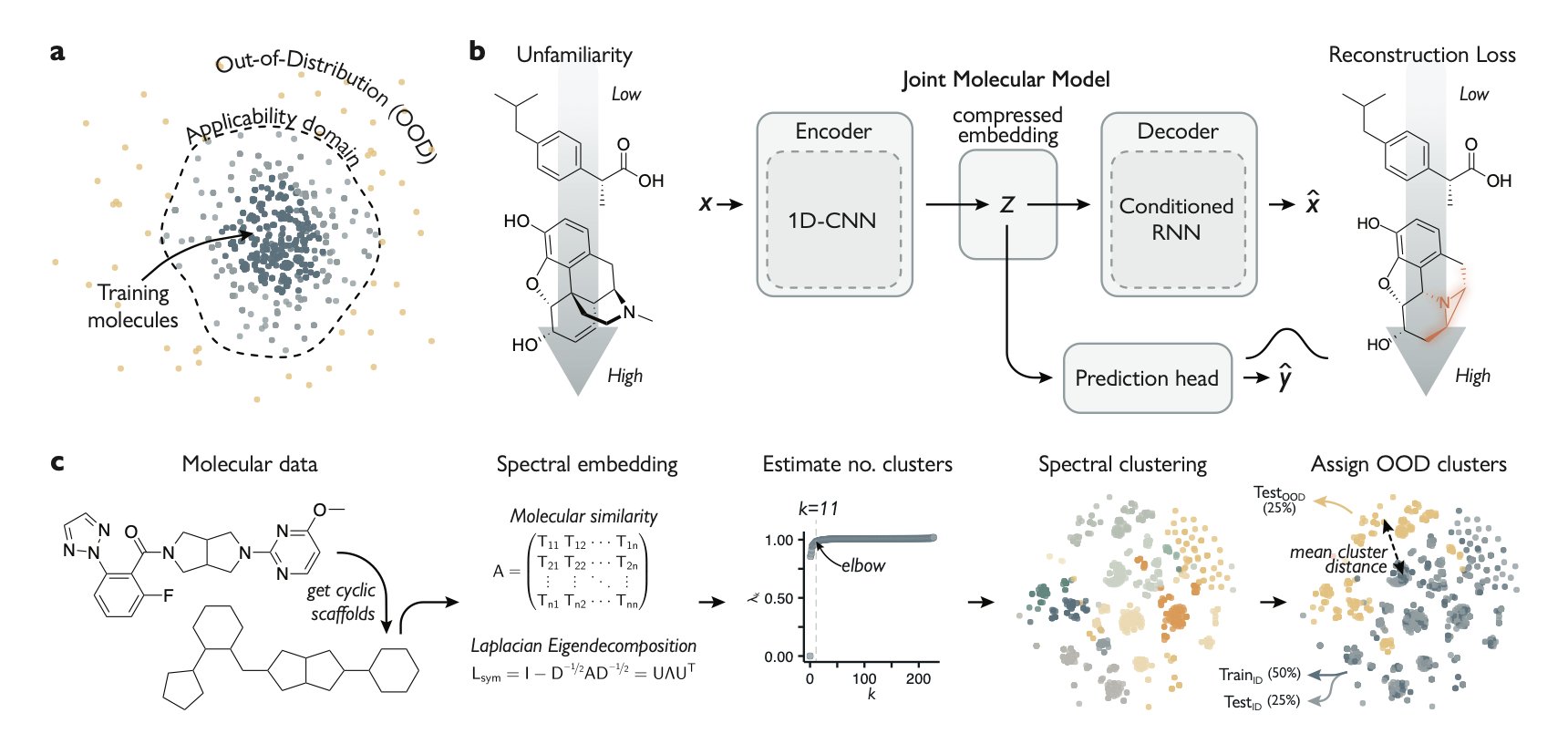
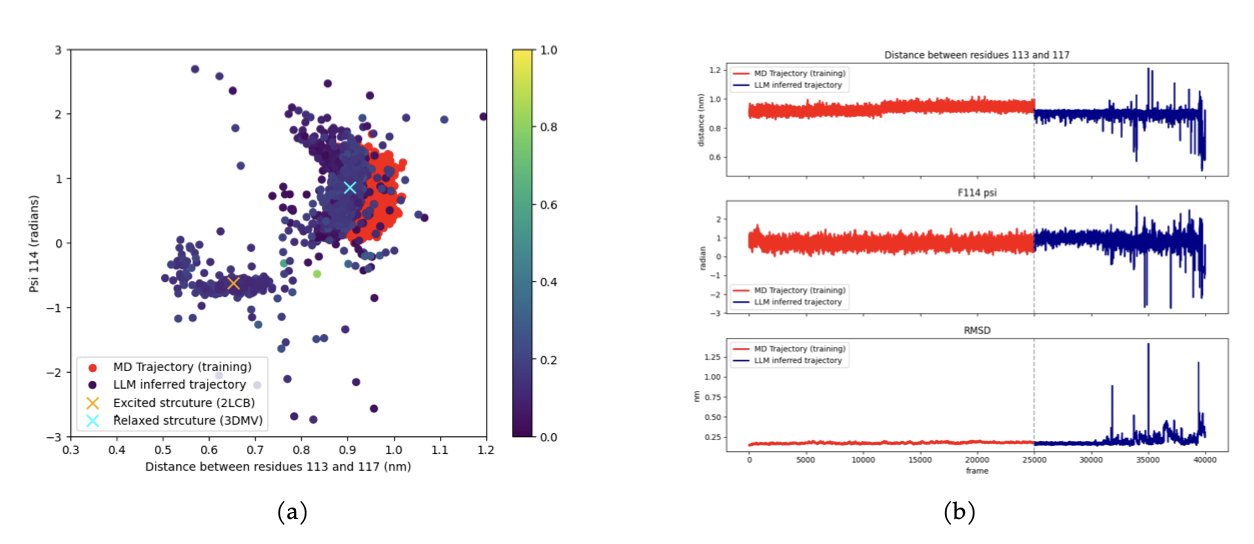
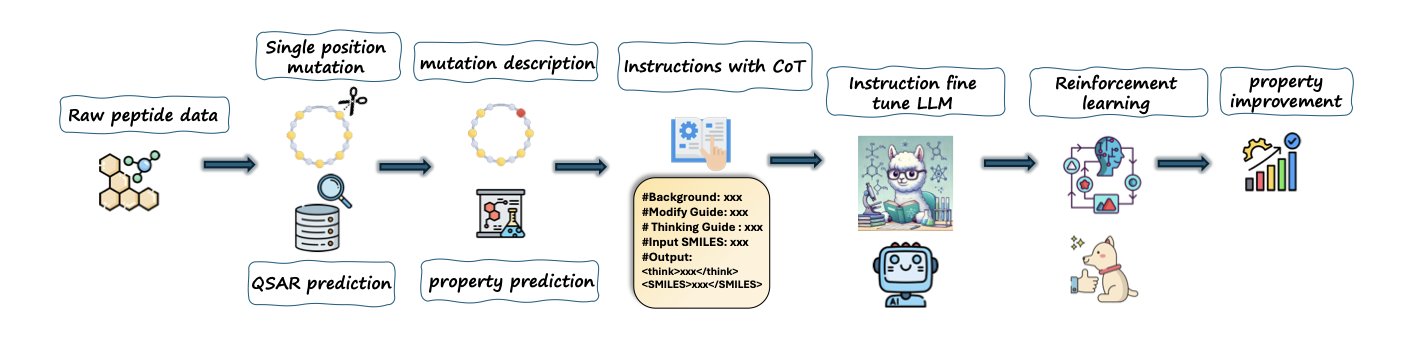
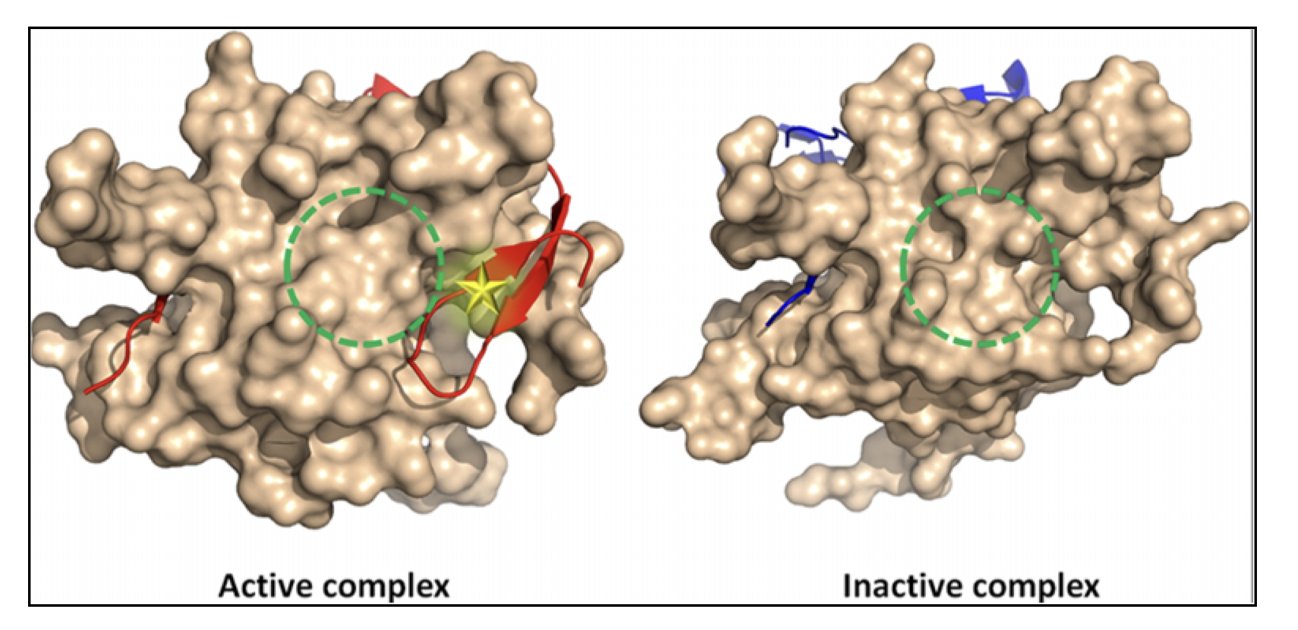
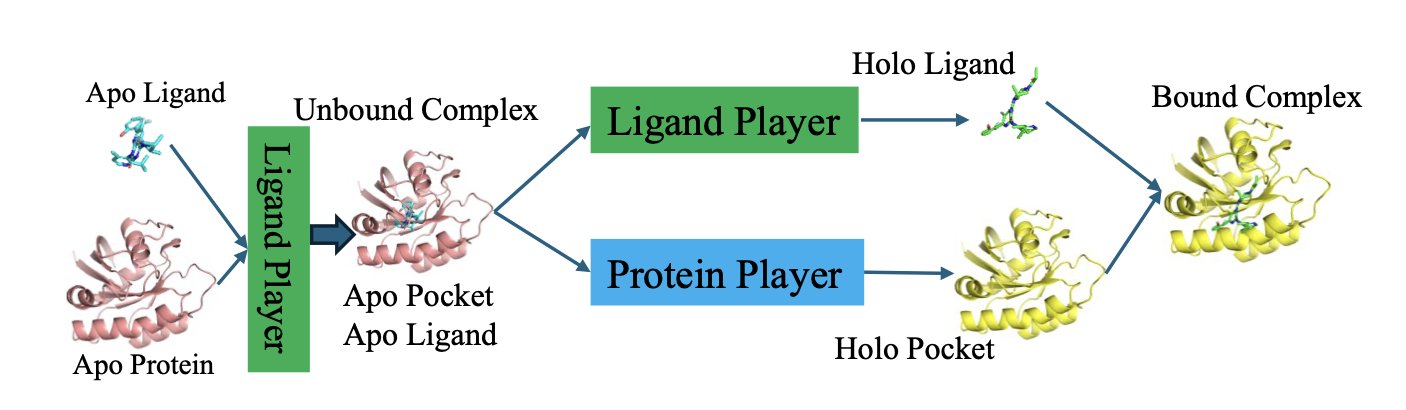
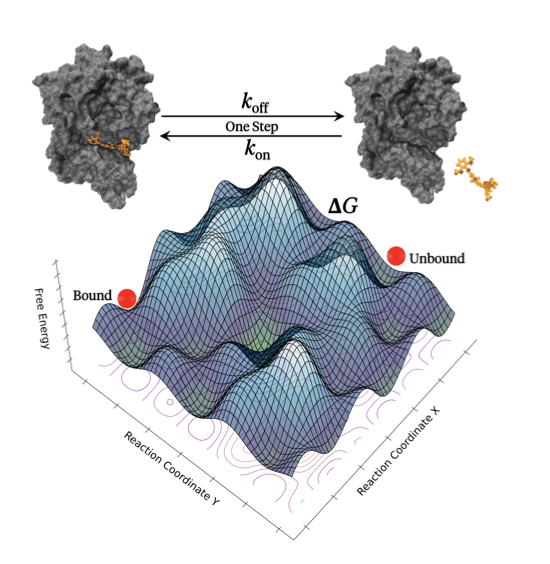
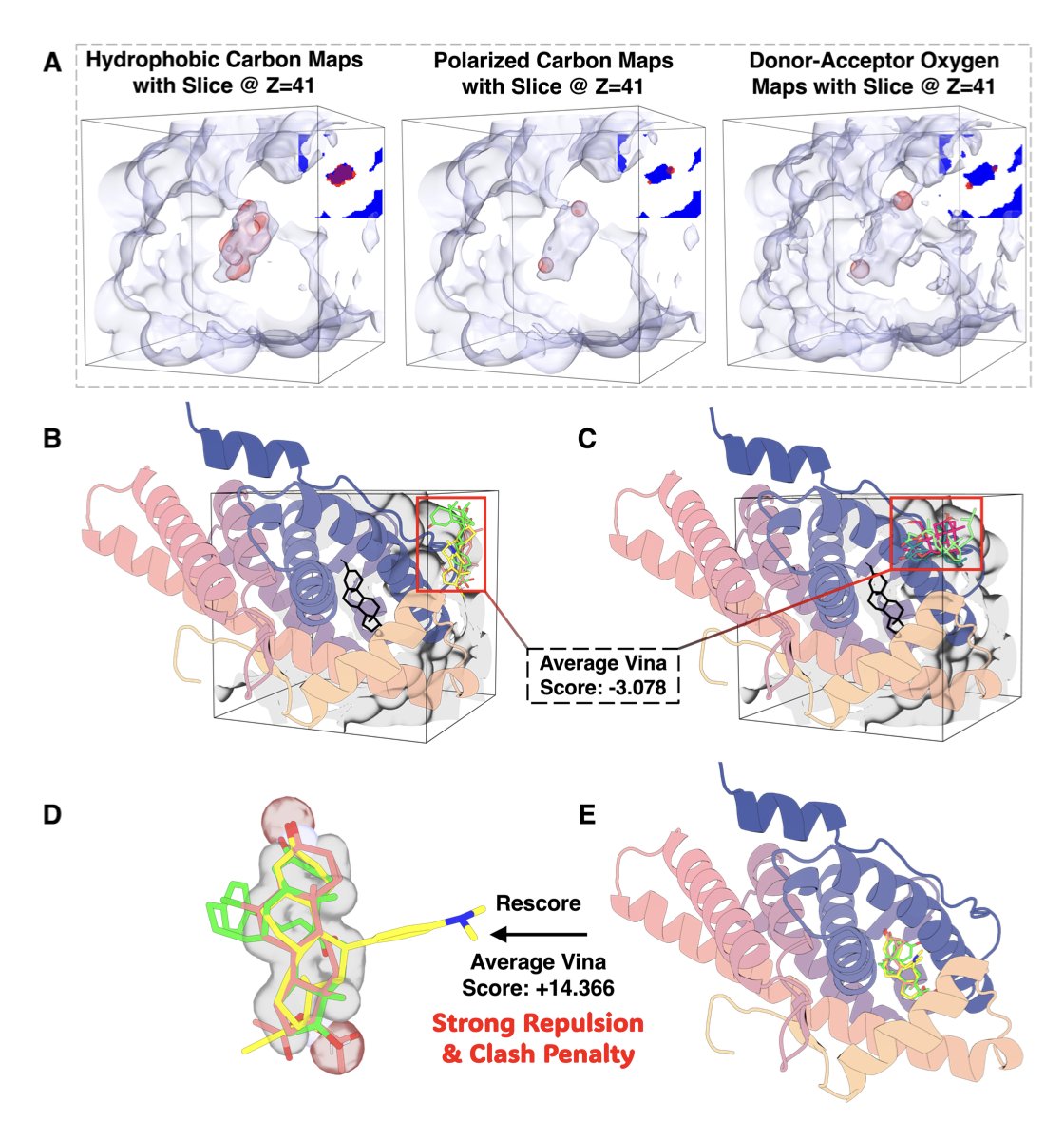
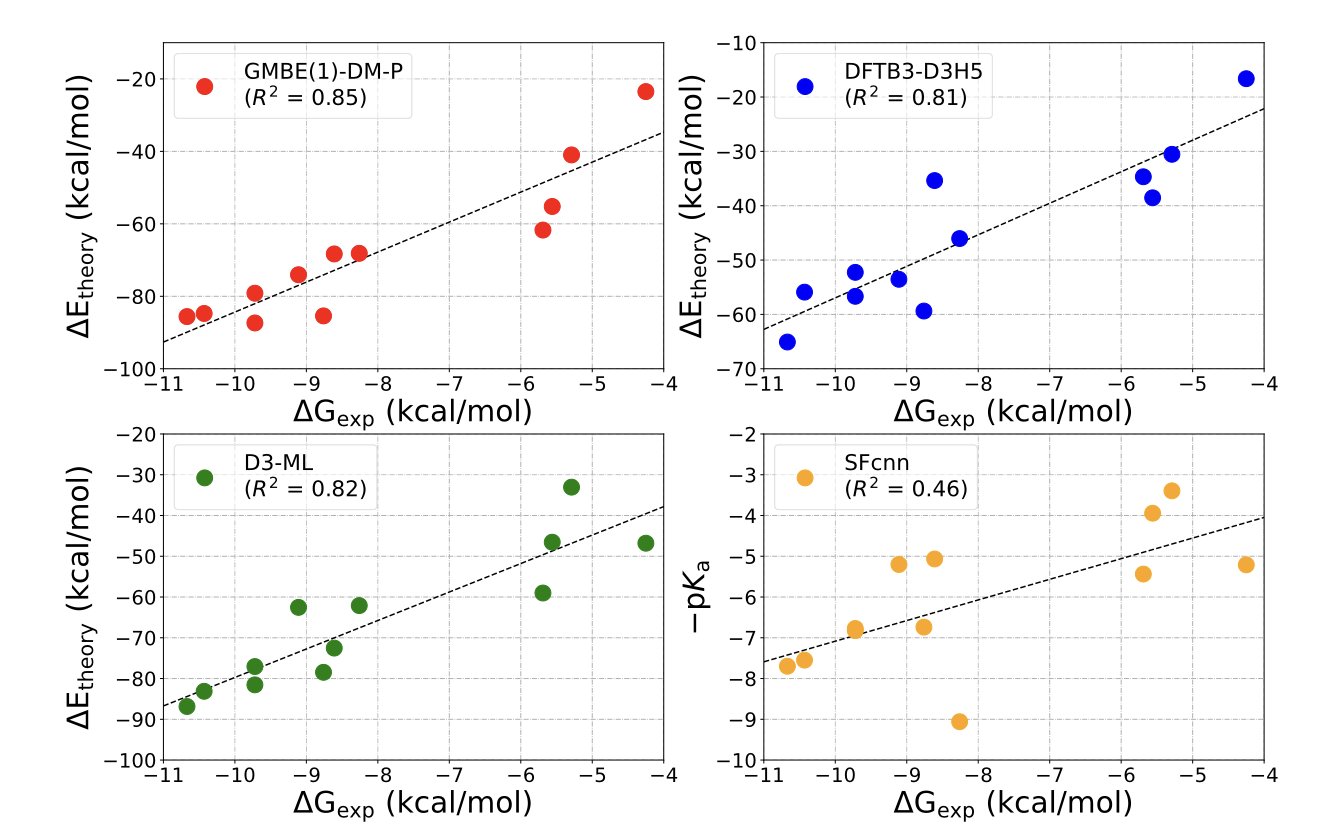
分子对接新博弈:让蛋白和配体对打
AI 药物研发
分子对接
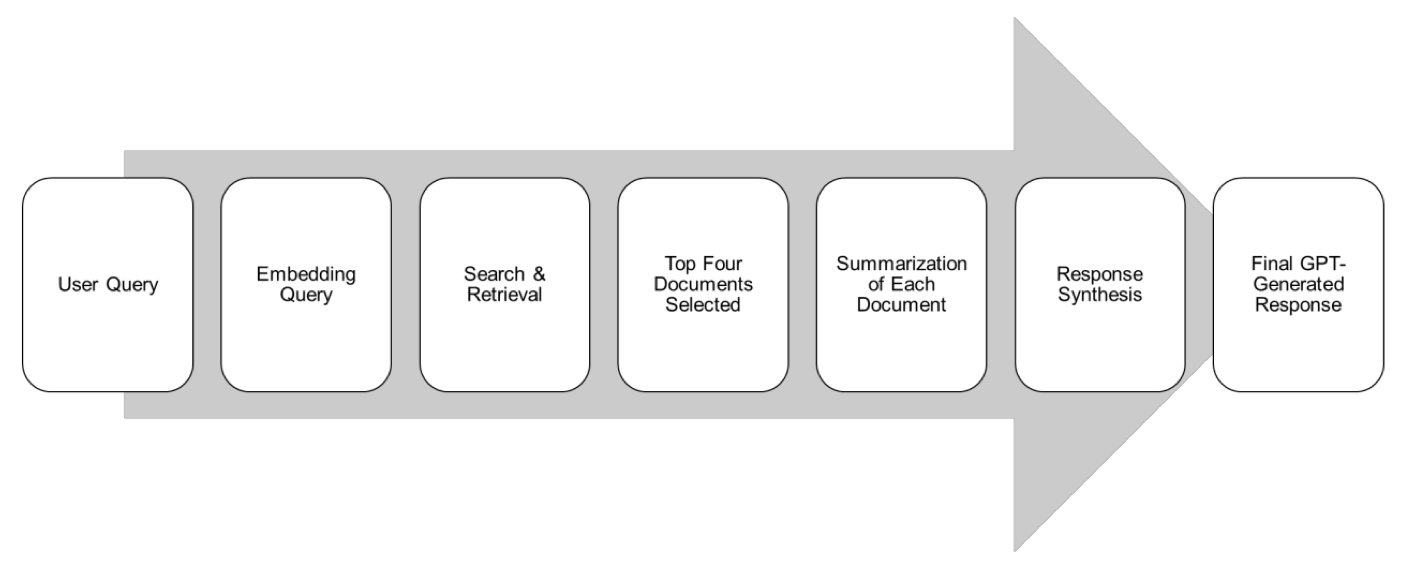
医学 NLP
化学反应预测
AI for Science
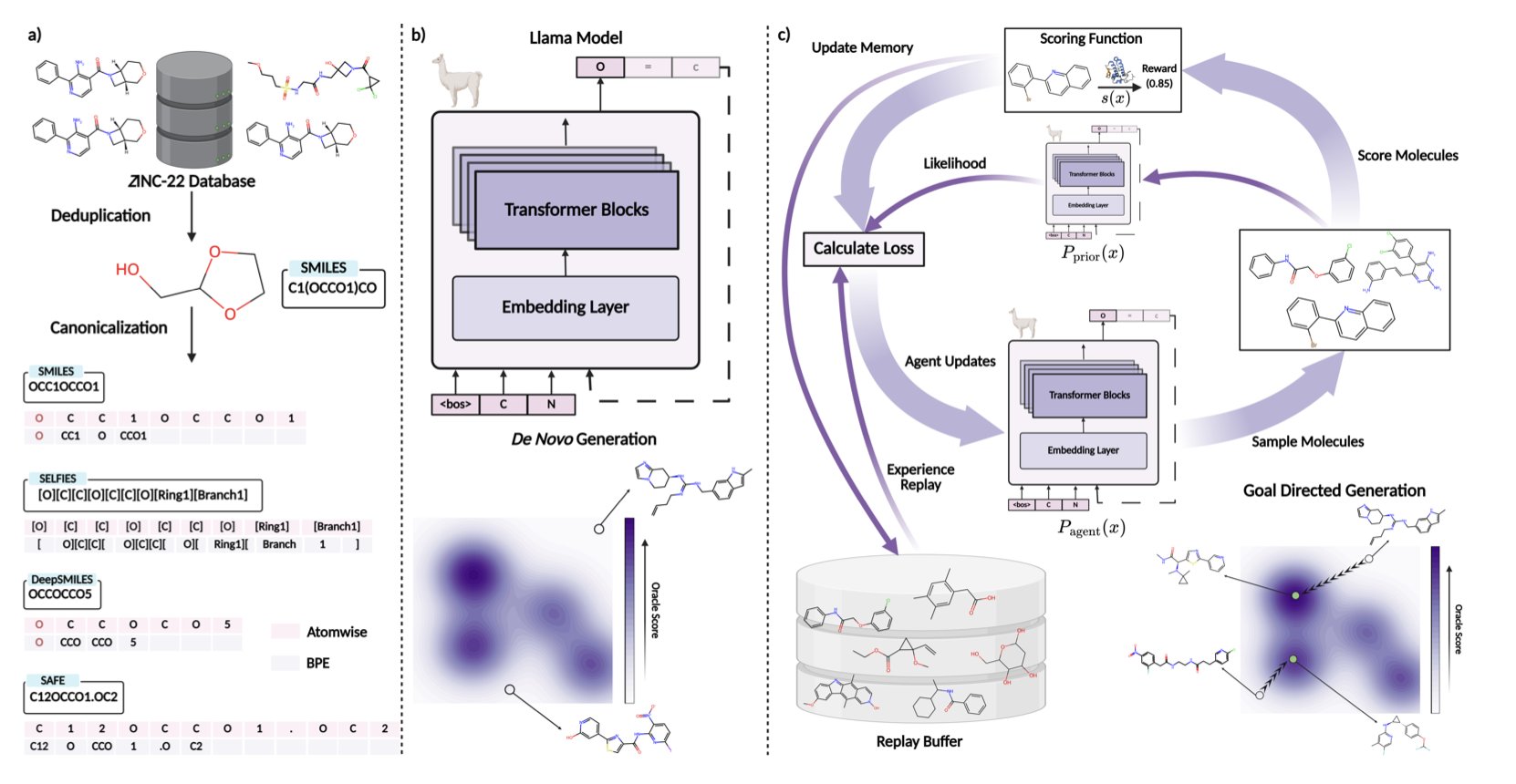
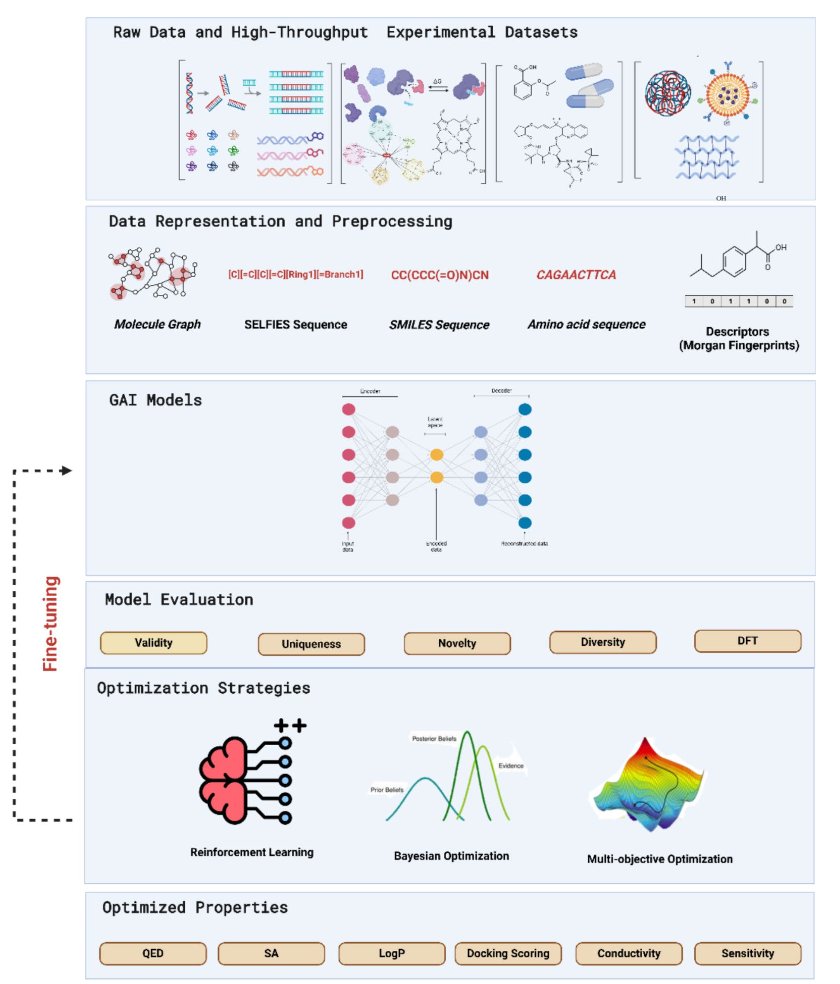
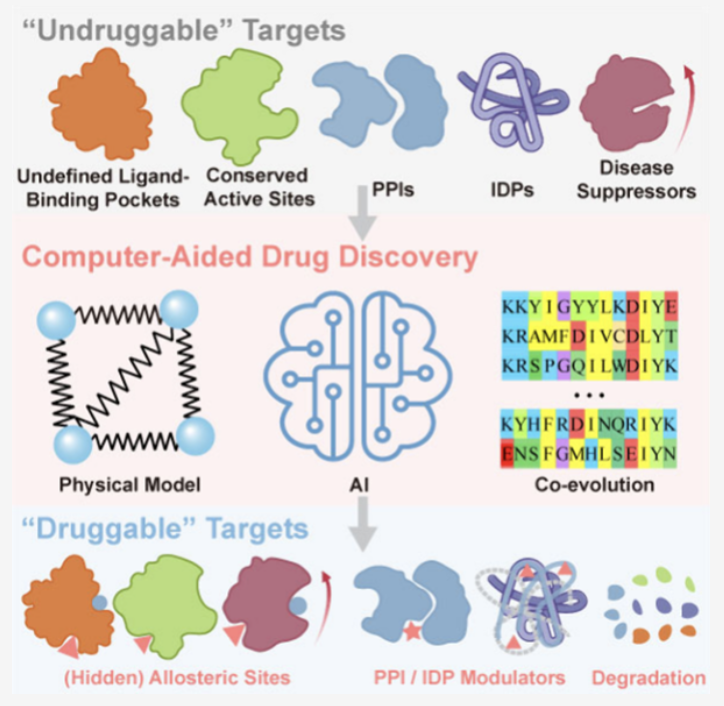
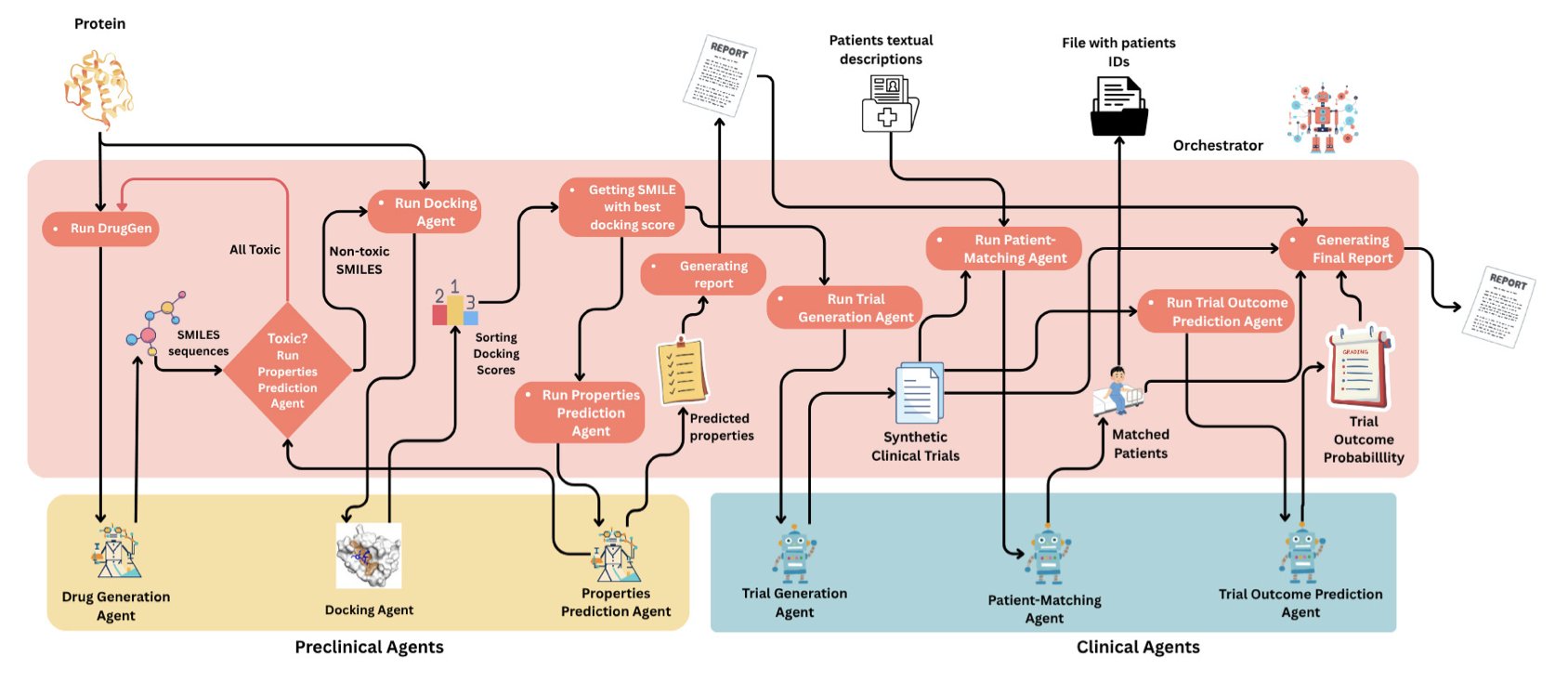
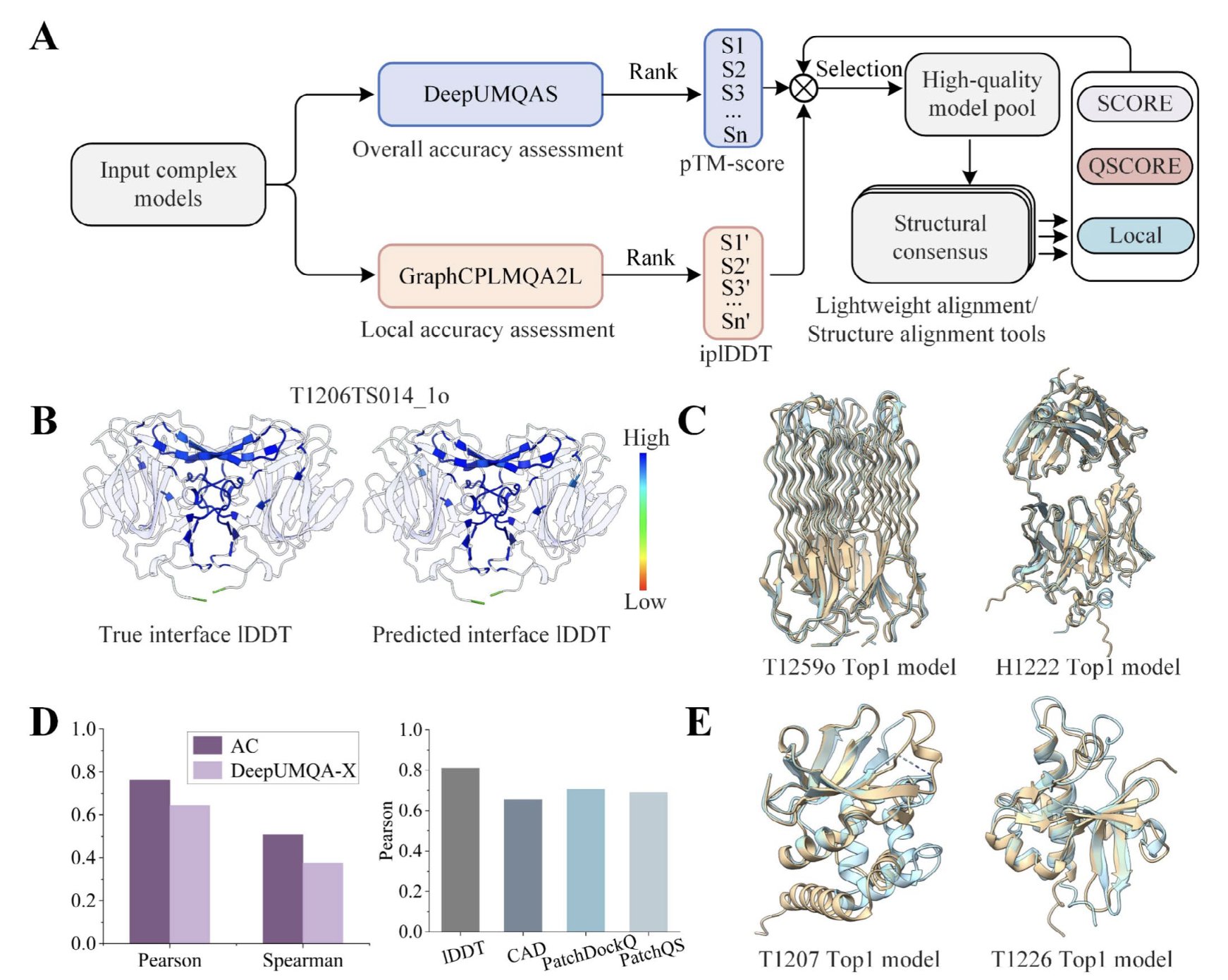
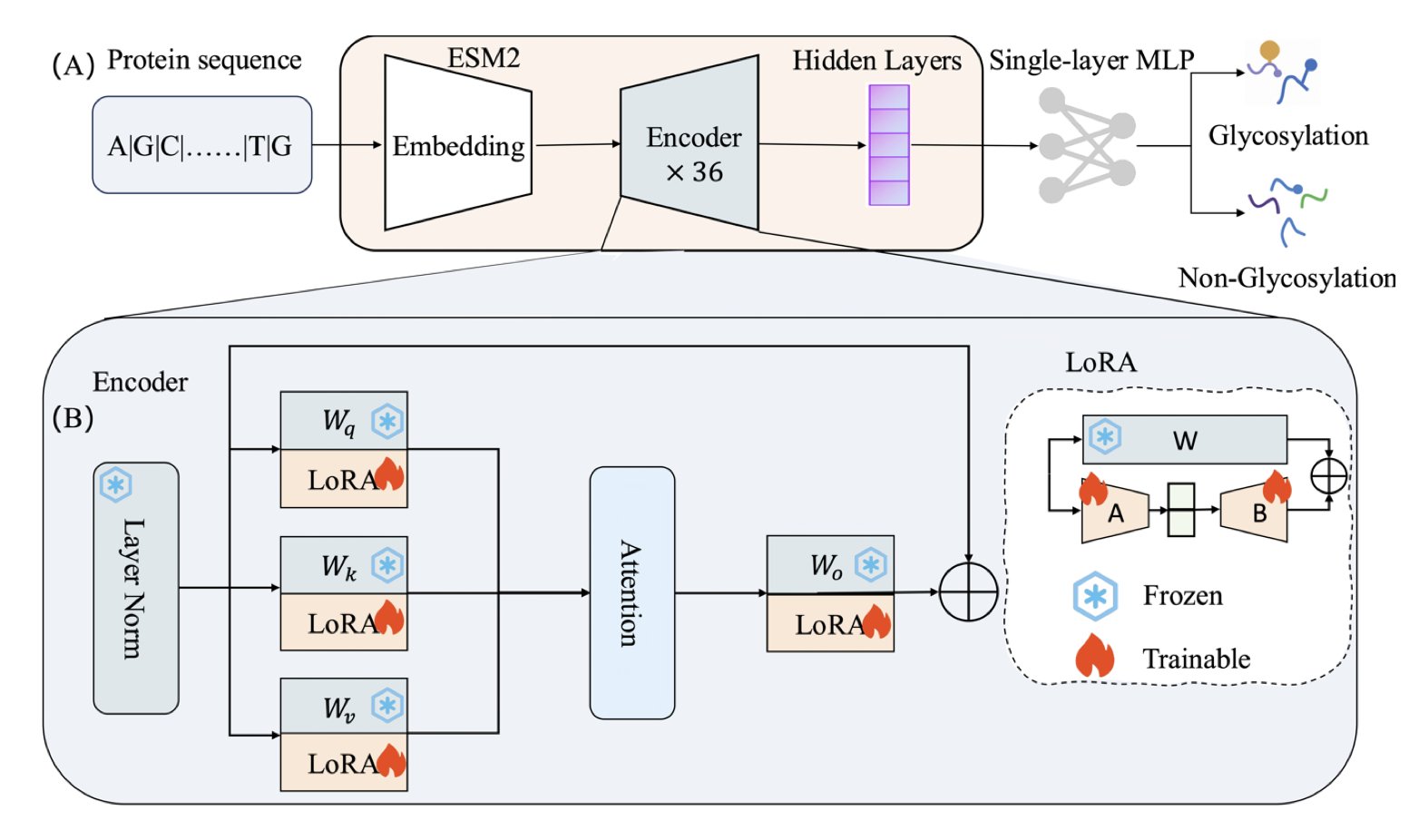
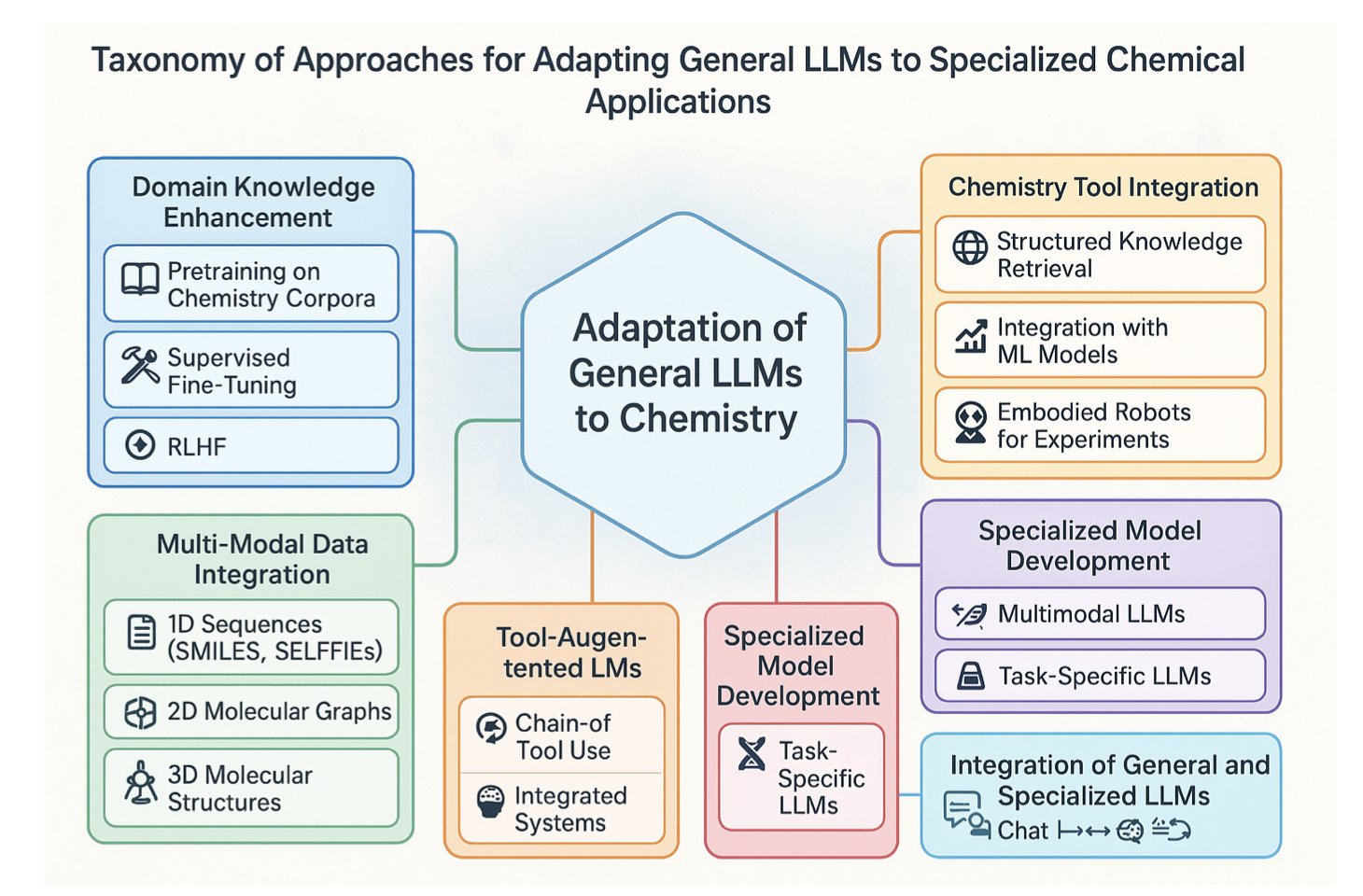
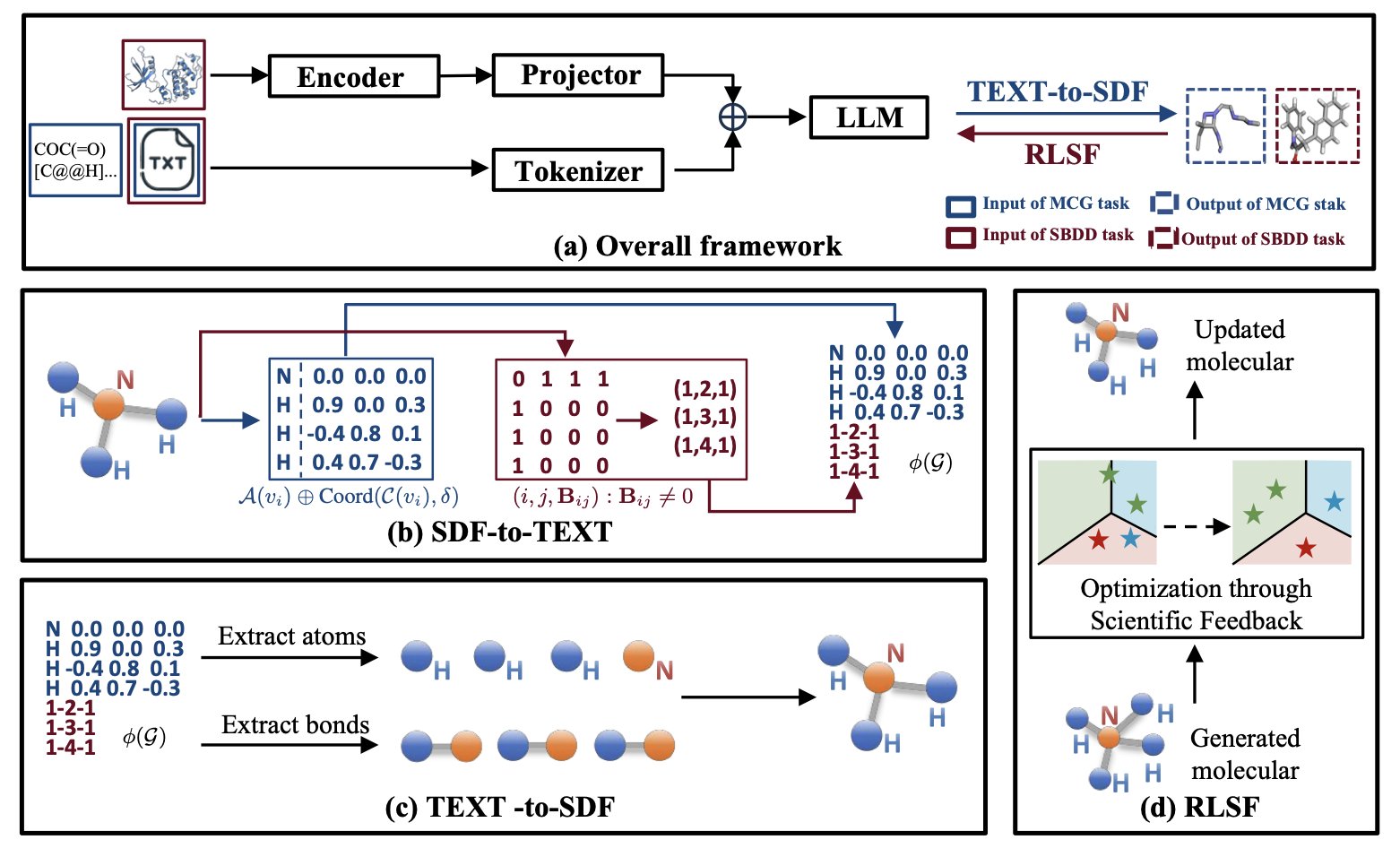
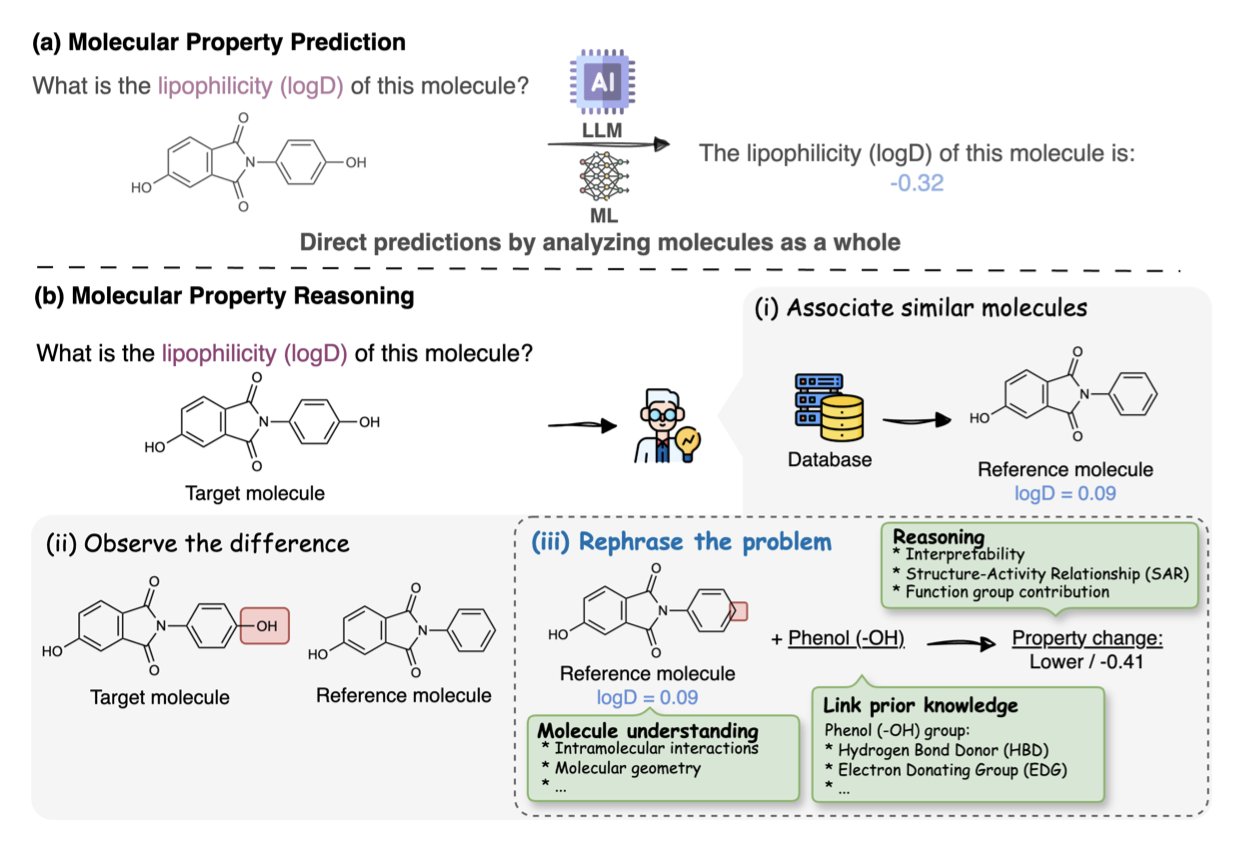
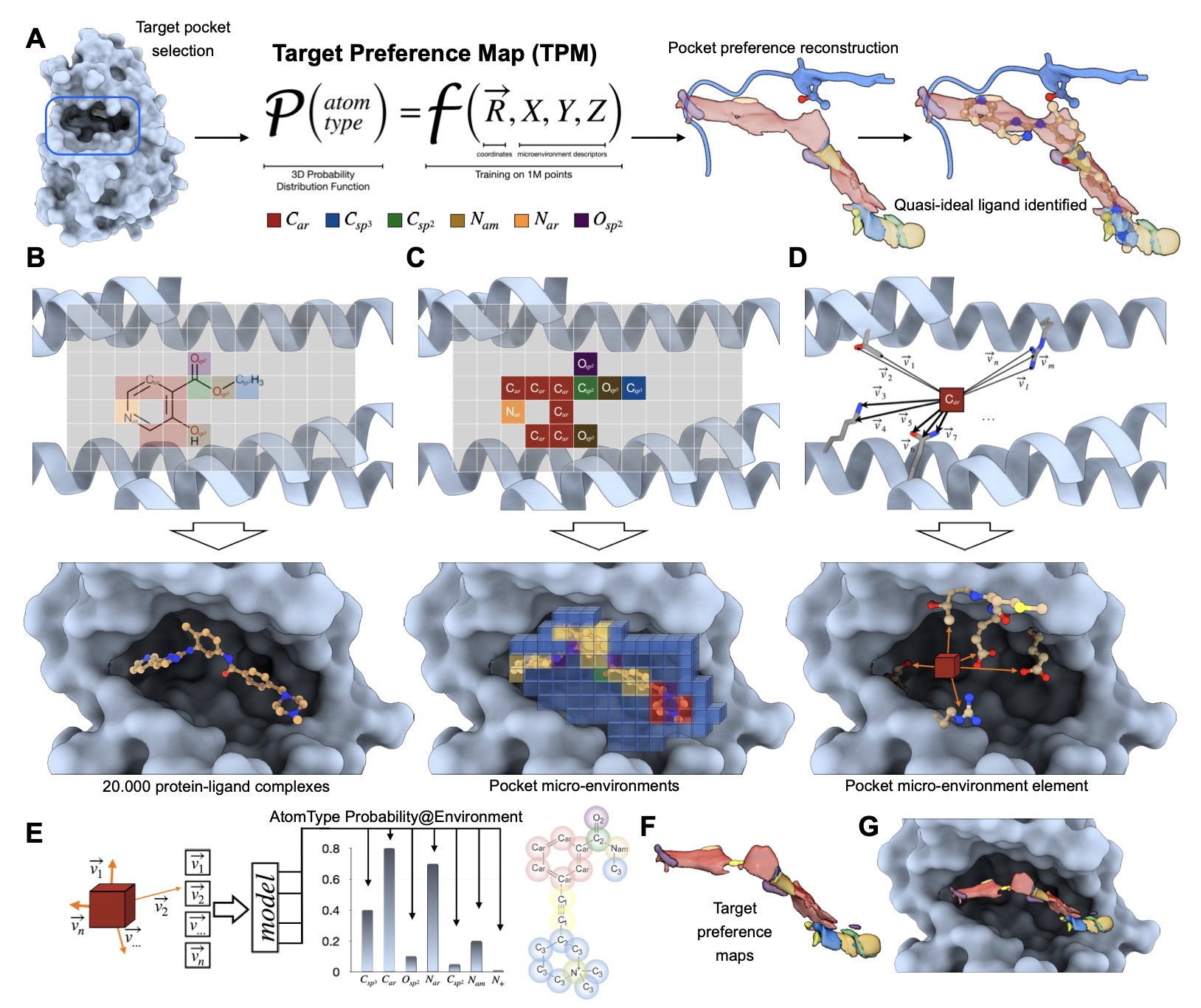
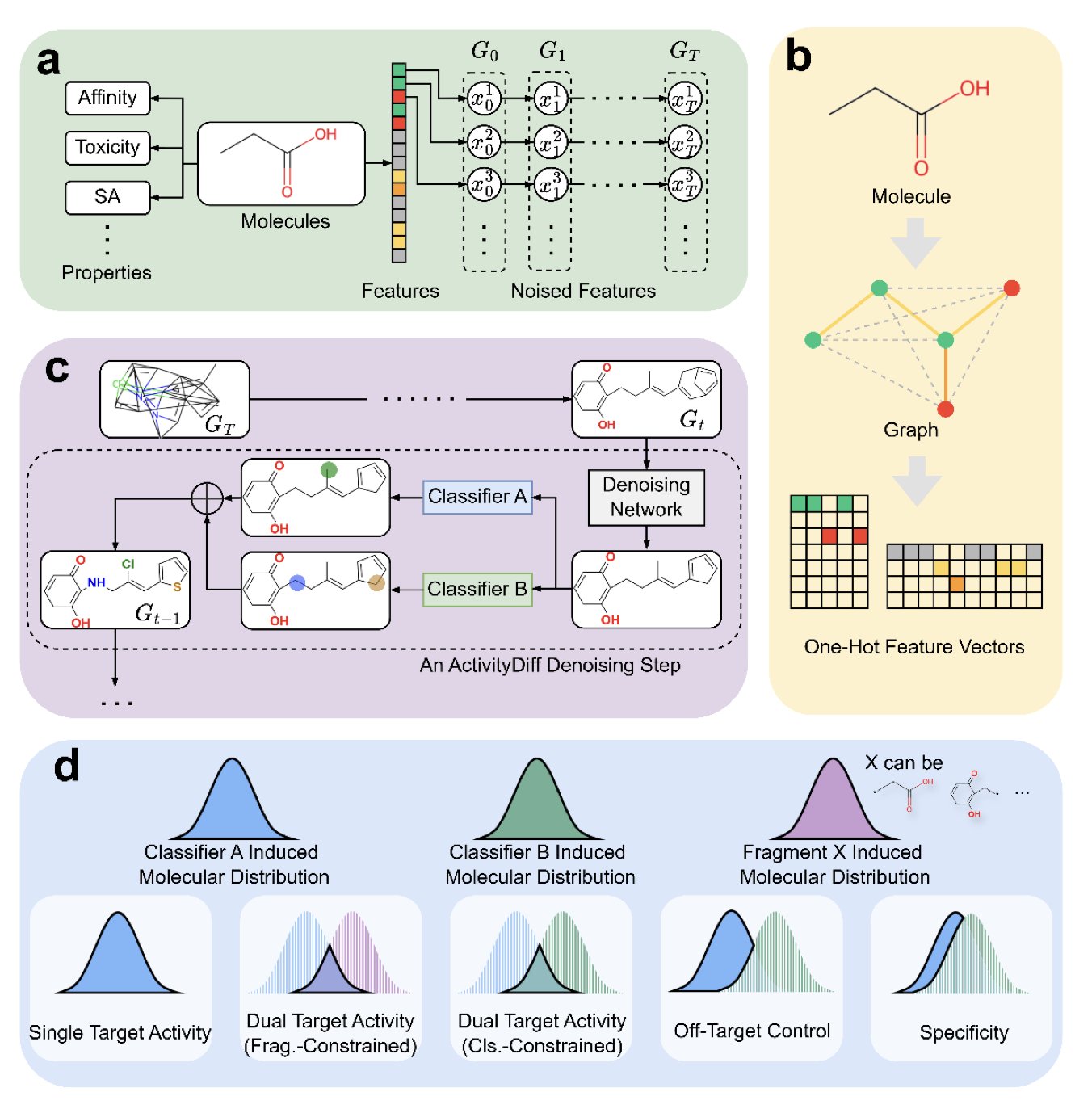
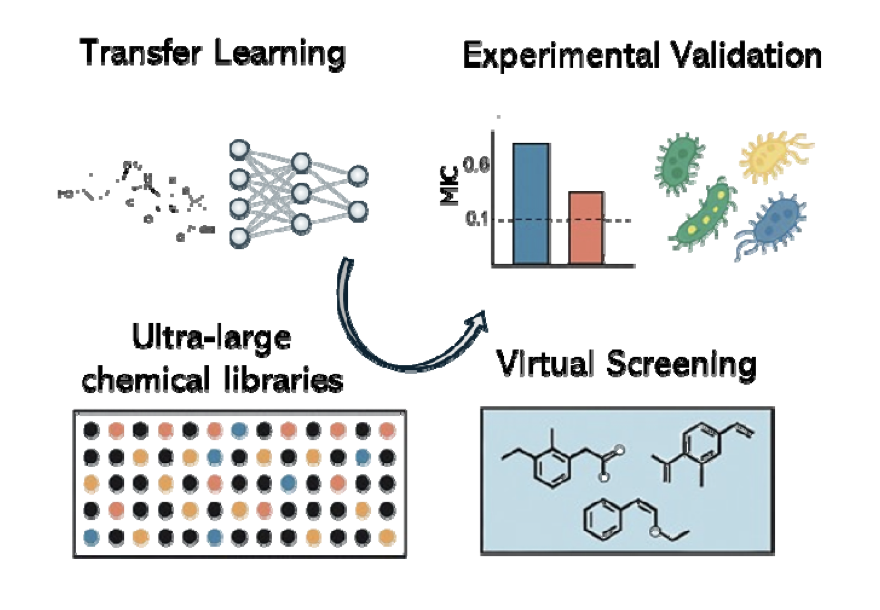
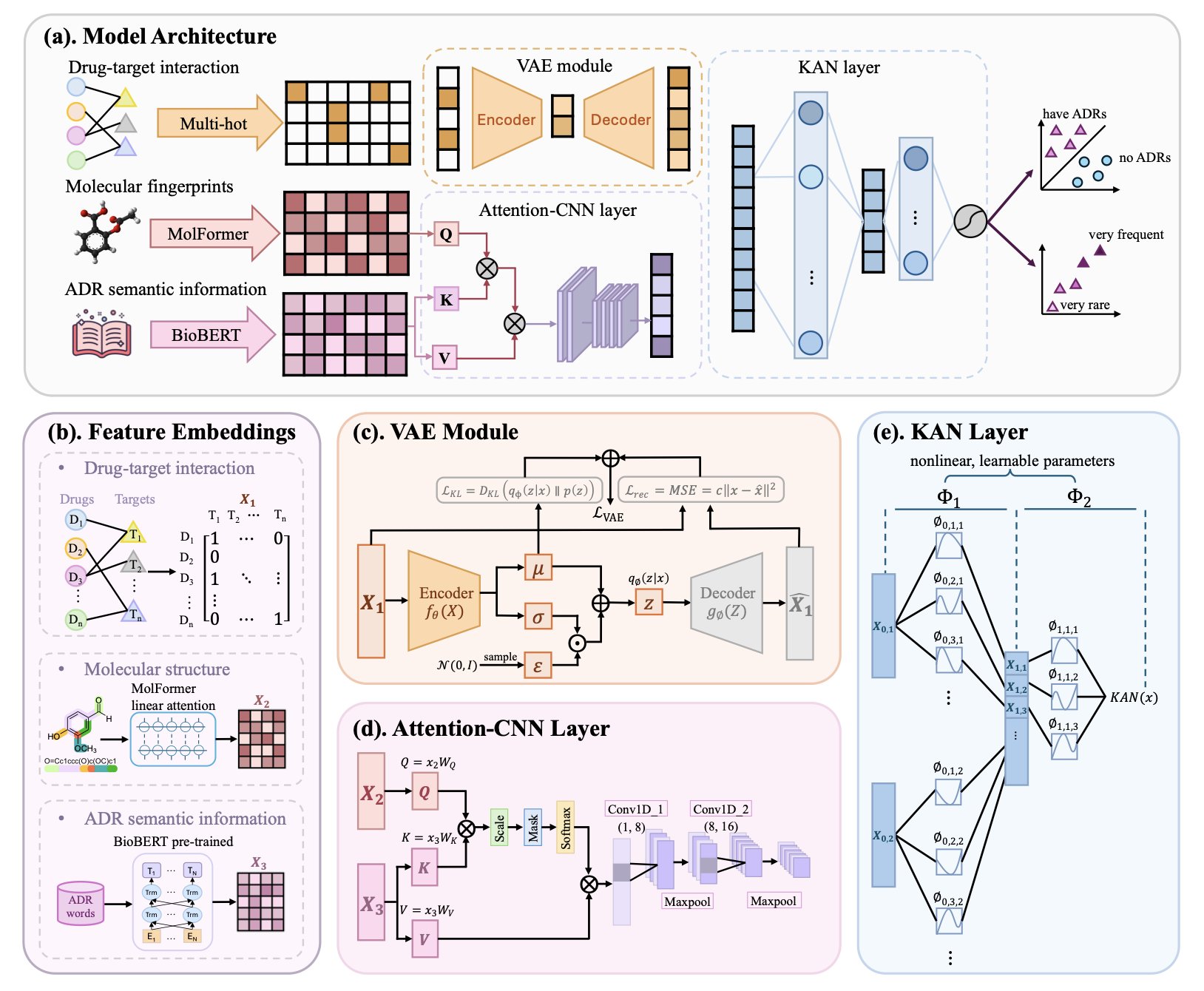
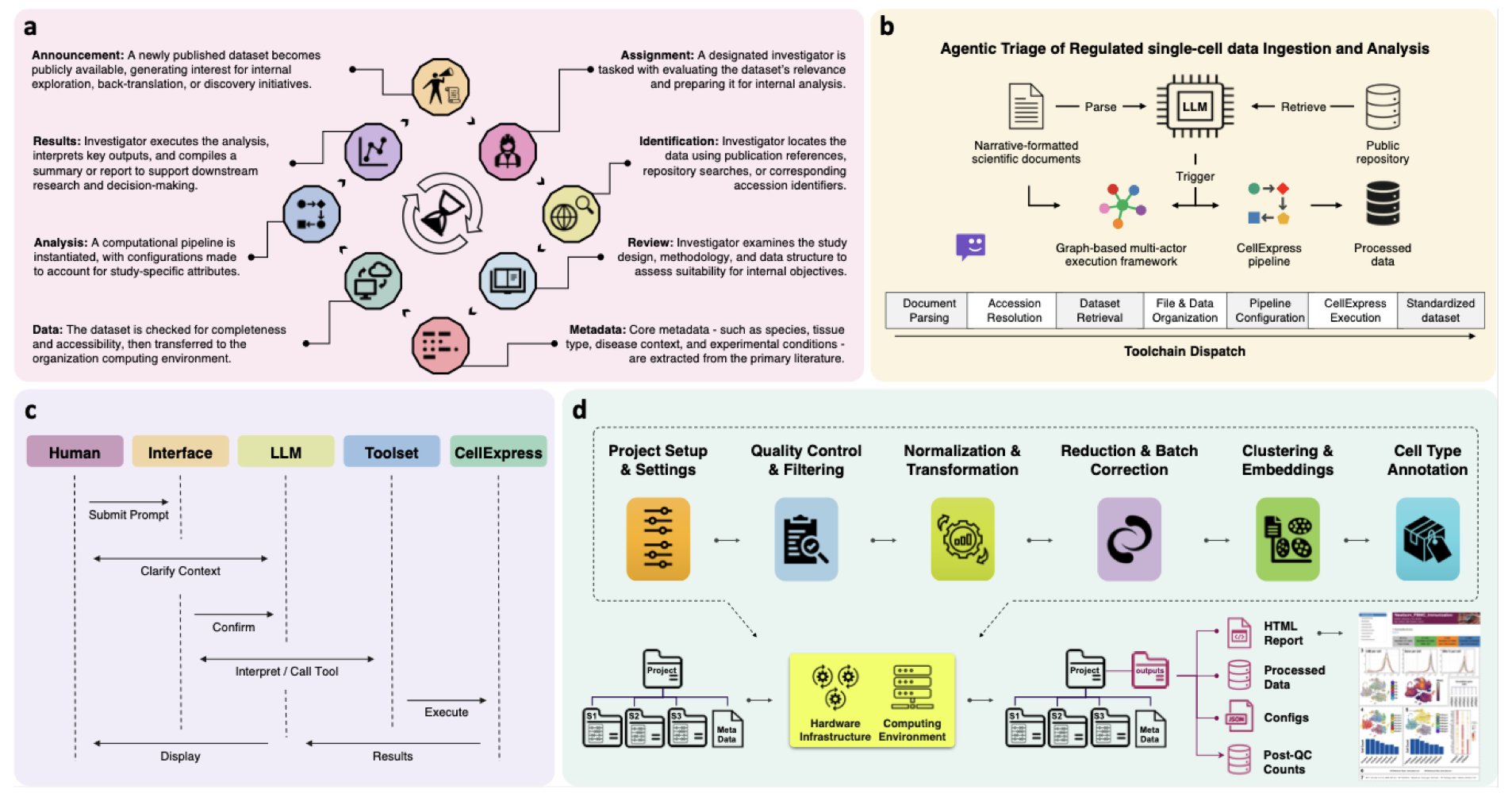
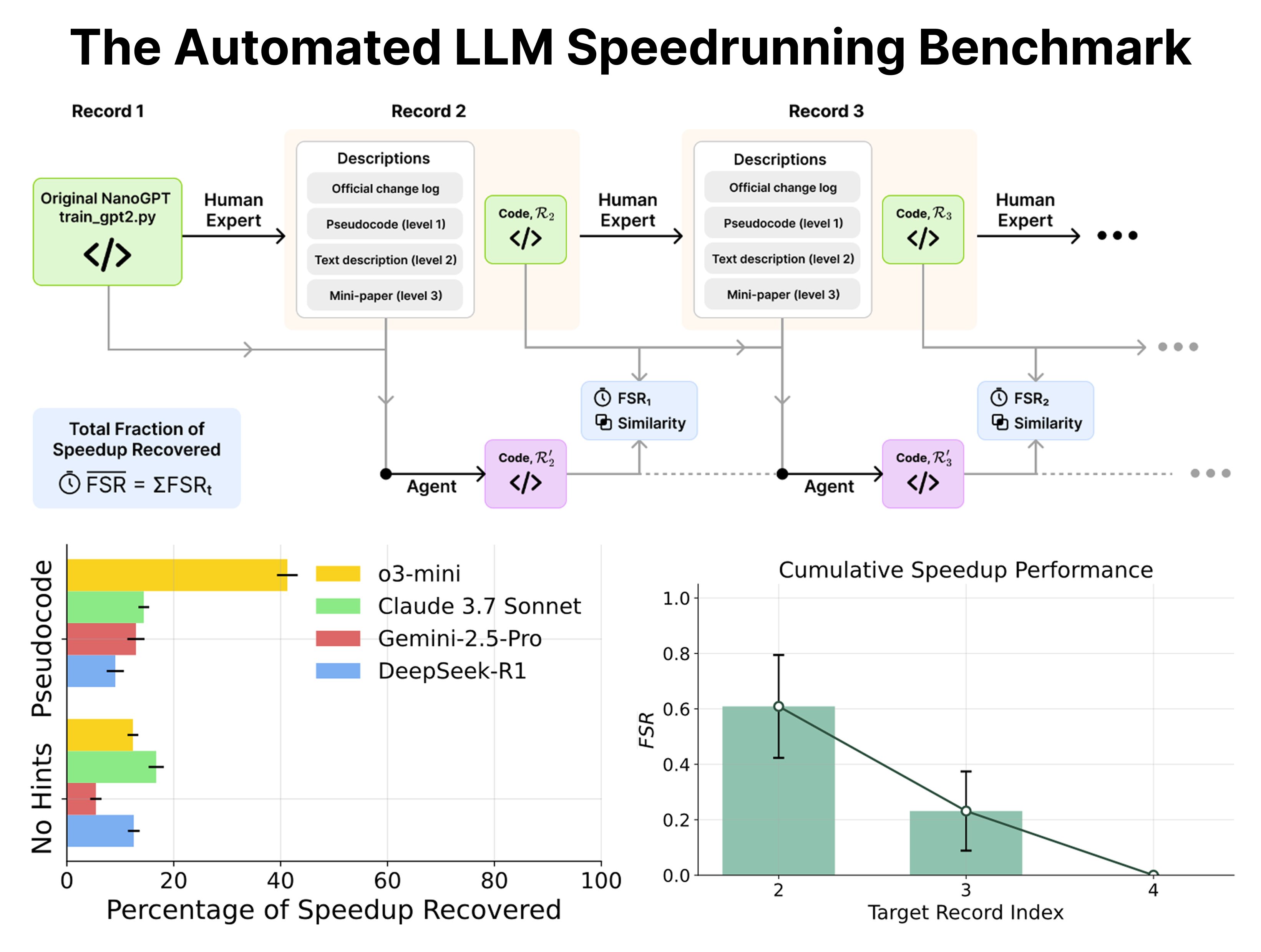
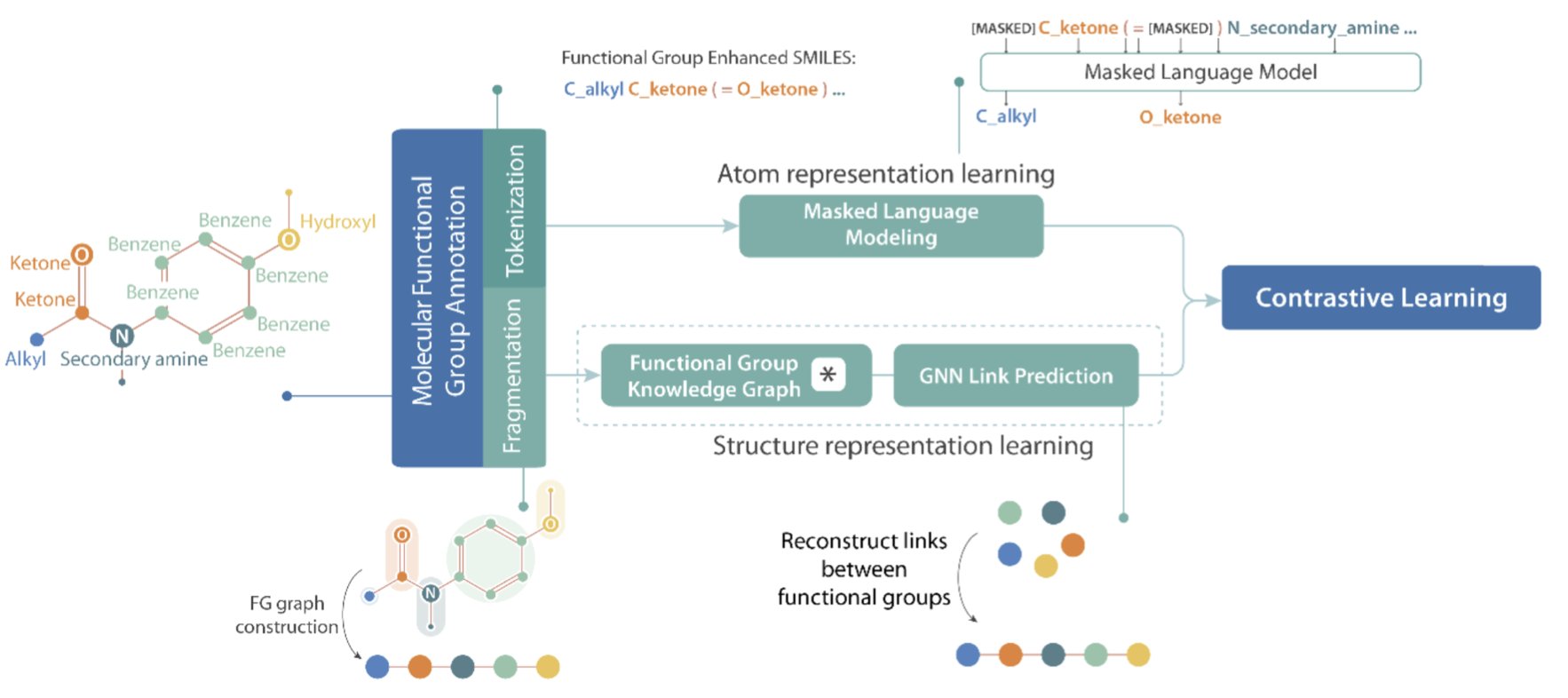
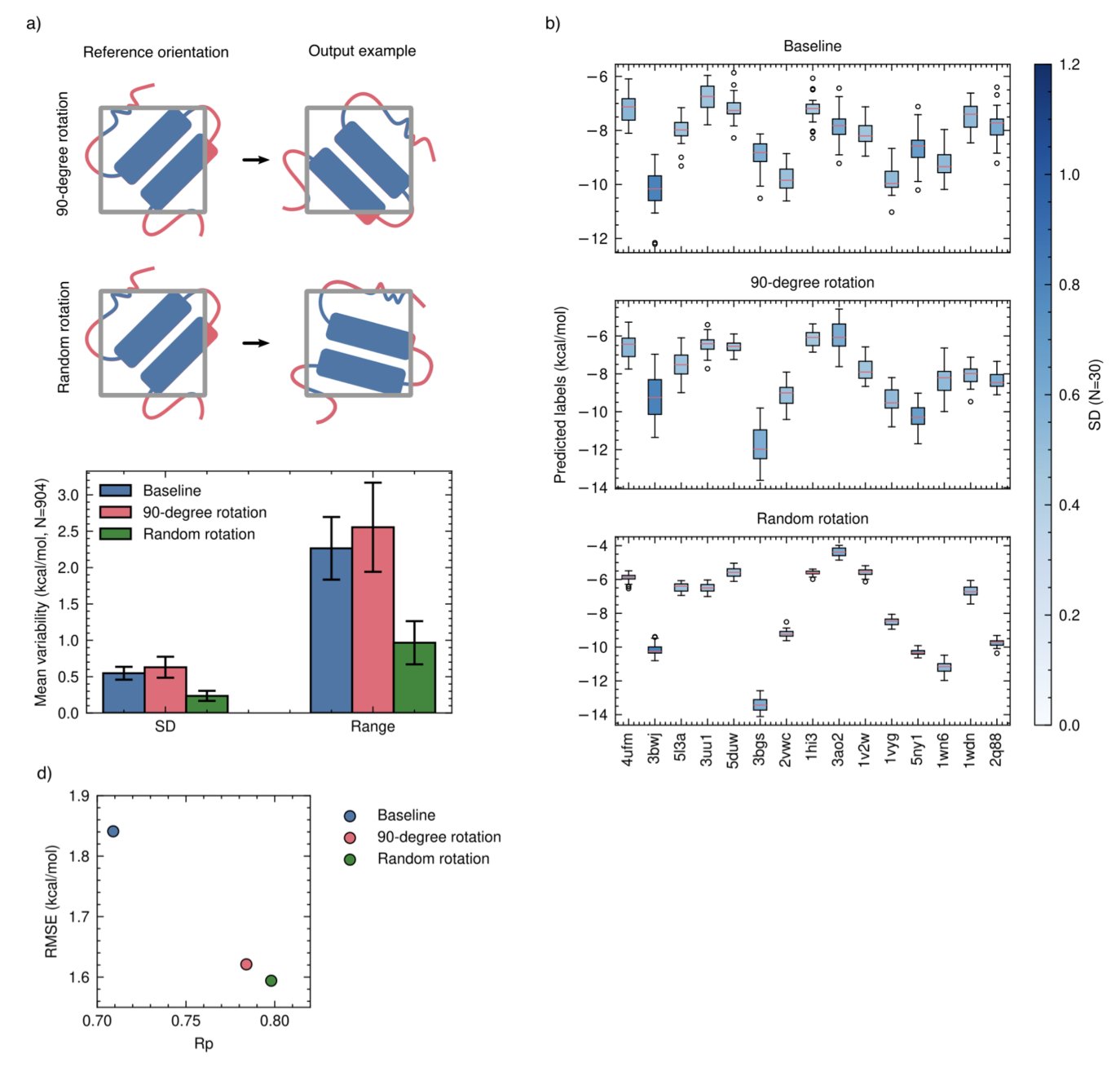
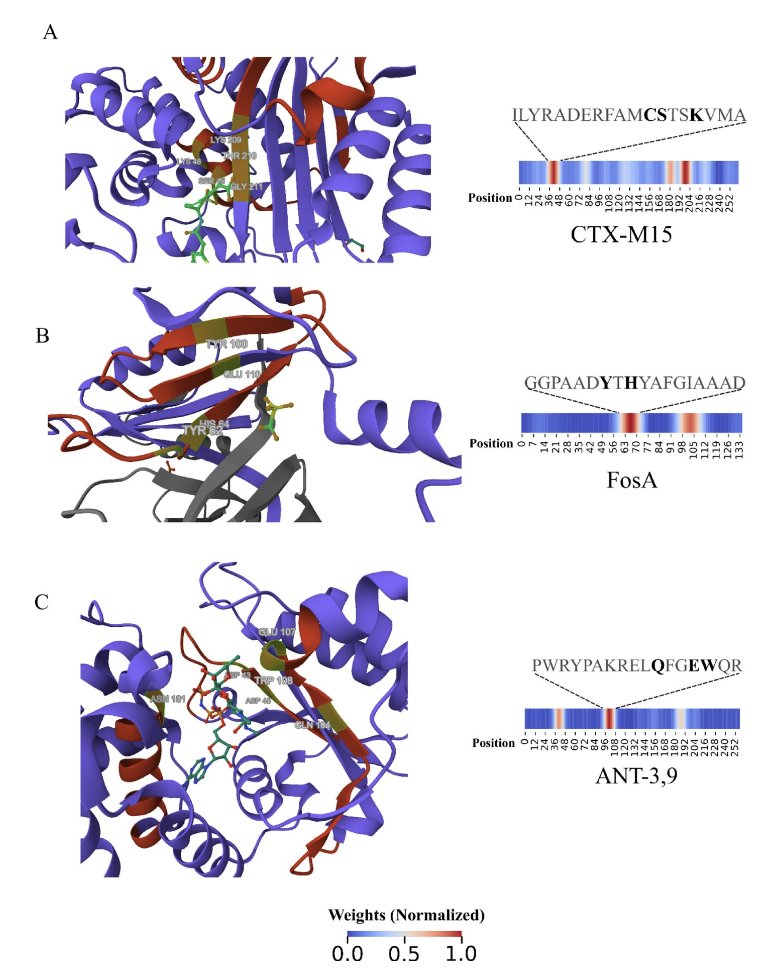
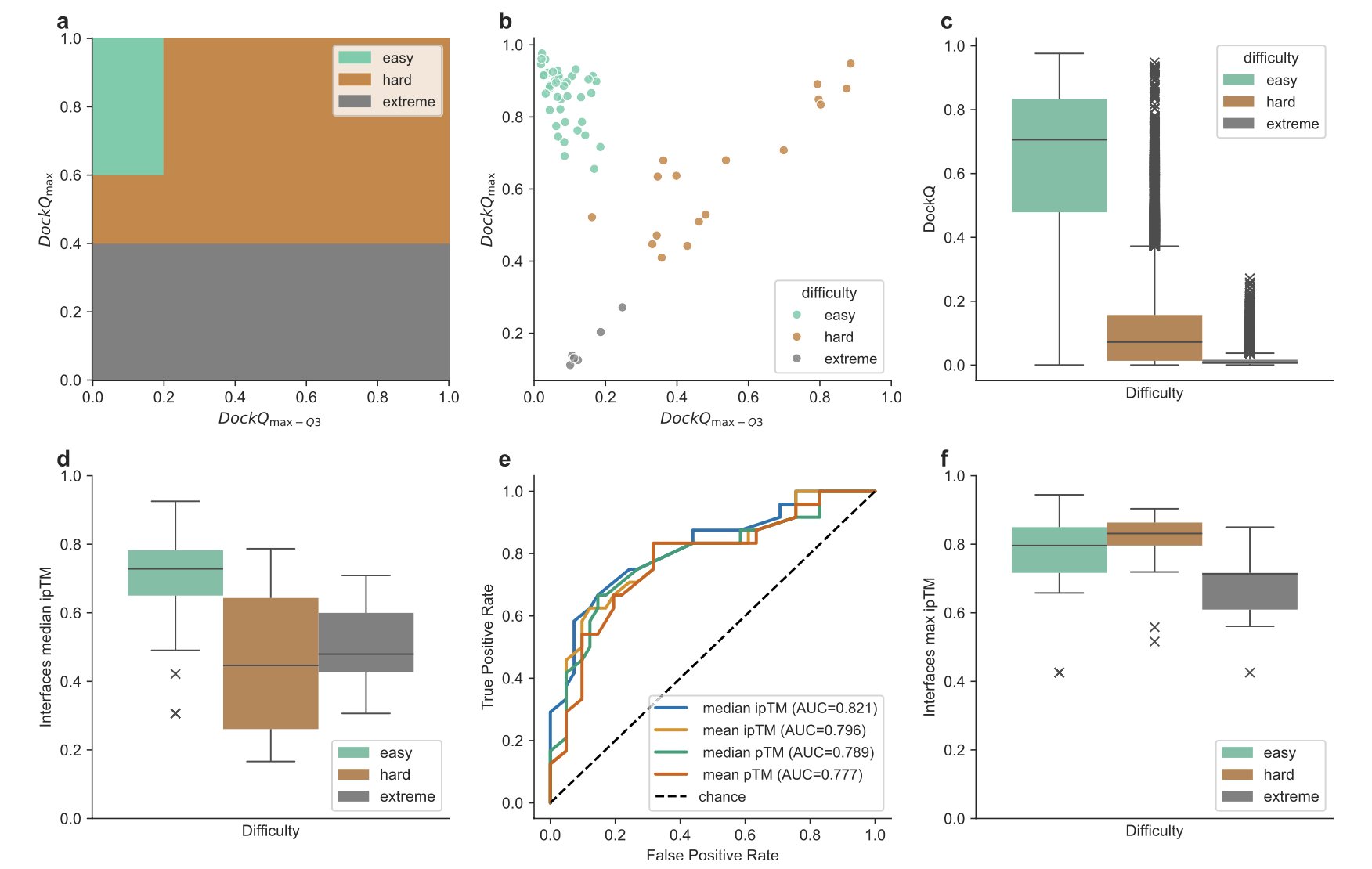
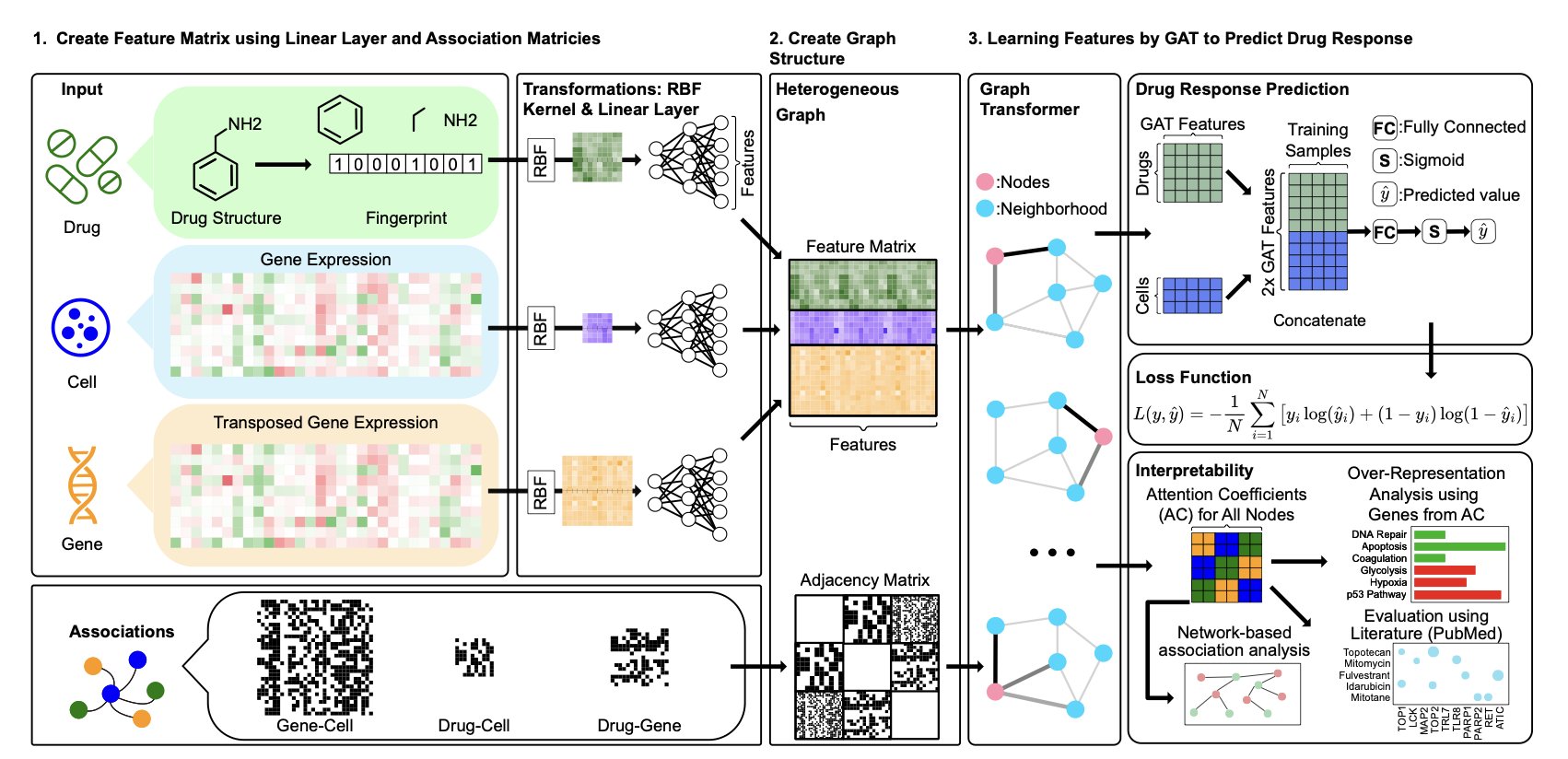
No matching items