
目录
- PETIMOT 是一个基于图神经网络的 AI 框架,它能直接从稀疏的静态 3D 结构中,快速、准确地推断出蛋白质复杂的动态运动,为药物研发中的构象分析提供了颠覆性的工具。
- 一项研究证明,一个简单、可解释并能评估自身预测置信度的 AI 模型,能够有效预测 hCA 抑制剂的选择性,并提供可行的分子设计思路。
- SiMGen 采用局部相似性方法,证明生成复杂分子无需海量数据,为药物设计提供了更灵活、可控的新思路。
1. PETIMOT:AI 预测蛋白动态,快到离谱
在药物研发中,蛋白质的动态至关重要。它们并非静止结构,而是在持续扭动、折叠。捕捉到那些能让药物分子结合的特定构象,是成功的关键。传统上,观察这些动态依赖分子动力学(MD)模拟,这一过程既慢又昂贵。
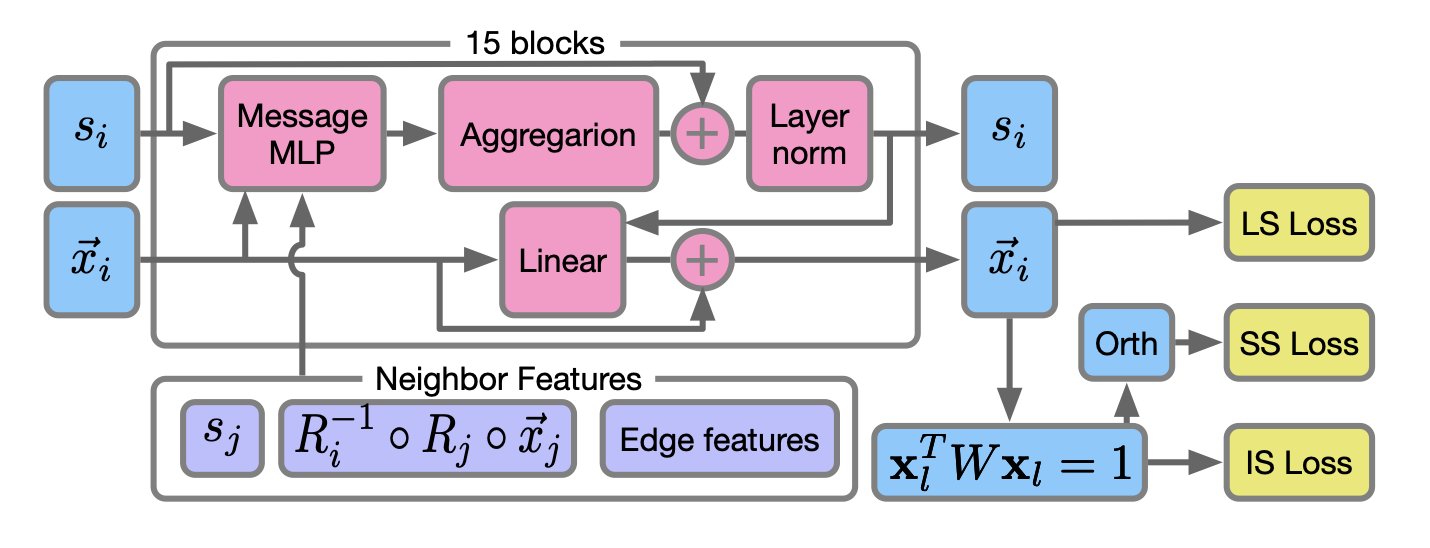
一个名为 PETIMOT 的新模型,能在几秒钟内完成这项任务。它本质上是解读蛋白质的 3D 结构图,并预测其动态变化。
PETIMOT 绕开了 MD 模拟对精确力场和海量计算的依赖。它的核心思路是,既然蛋白质的运动模式在同源蛋白间具有共通性,模型可以直接从蛋白质数据库(PDB)中海量的已知静态结构里,学习这些运动的内在规律。
为实现这一点,研究者使用SE(3) 等变图神经网络架构。这种架构将物理规律编码进模型:无论如何旋转或平移蛋白质,其内部运动模式保持不变。这让 AI 学习得更快、更准。
PETIMOT 还建立在蛋白质语言模型(Protein Language Models, PLM)的基础上。PLM 已能从序列数据中理解氨基酸如何构成蛋白质,PETIMOT 则更进一步,教 AI 如何将这些静态的氨基酸残基组织成动态的构象变化。
模型的核心是学习一个「运动可能性空间」(技术上称为位置协方差矩阵的本征空间)。它不预测一个蛋白会精确移动到某个点,而是描绘出其所有可能的移动方向与范围。这样,模型归纳出了一套通用的运动规律,而非记忆特定蛋白的动作。
结果是,该模型处理整个测试集仅用 16 秒,而其他 AI 方法或传统物理方法需要数小时。
这一进步将改变药物研发的两个关键环节。
首先是寻找隐蔽口袋(cryptic pockets)。许多蛋白质的活性位点在常规状态下是关闭的,仅在特定动态构象下才会短暂开启一个可结合的口袋。过去依赖 MD 模拟等待口袋出现,效率很低。PETIMOT 能快速生成成百上千种高概率构象,供对接软件筛选,显著提高了找到这些口袋的机会。
其次是别构调节剂的开发。别构药物结合在活性位点之外,通过改变蛋白质整体形状来发挥作用。PETIMOT 预测的蛋白质全局运动模式,为寻找潜在的别构位点提供了一张精确的地图。
PETIMOT 也有其局限。它依赖已有的 3D 结构数据,无法处理结构未知的蛋白质。同时,它预测的是「运动的可能性」,而非在特定条件(如结合候选药物后)下的确定结果。
因此,它最合适的定位是一个高效的「构象搜索引擎」,用于快速产生大量可靠的假说,再由 MD 模拟或生物实验等方法进行验证。
2. AI 预测蛋白选择性,并解释为什么
药物发现的核心挑战之一是「选择性」:如何让药物分子只攻击目标靶点,而不误伤结构相似的「亲戚」蛋白。
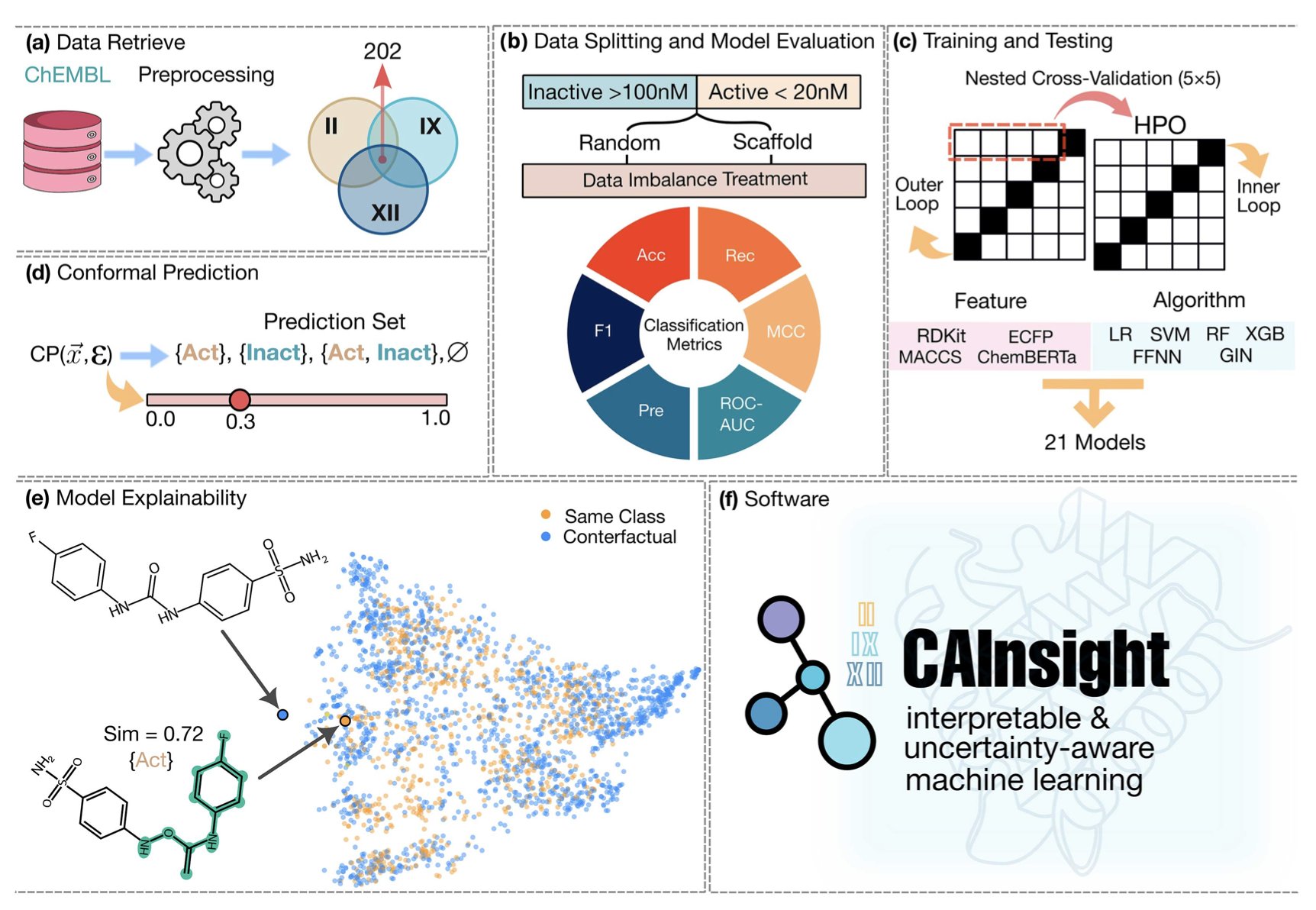
碳酸酐酶 (Carbonic Anhydrase, hCA) 家族就是这样一个例子。为了治疗癌症,研究者希望抑制 hCA IX 和 XII 亚型。然而,人体内广泛存在功能重要的 hCA II 亚型,一旦被错误抑制,就会引发副作用。因此,设计出能精准区分这些亚型的分子至关重要。
过去,研究者常使用深度神经网络解决此类问题。这些模型虽然能给出预测,却像一个黑箱,无法解释其预测的依据。一个只告诉你「这个分子选择性好」却不说明原因的模型,对需要决定下一步合成方案的药物化学家帮助有限。一篇新论文提供了一套更符合化学家思维方式的工具。
简单模型胜出
研究者比较了多种机器学习模型,从简单到复杂。
结果,表现最好的并非复杂的深度学习网络,而是一个经典方法:支持向量机 (Support Vector Machine, SVM) 结合 化学指纹 (Extended-Connectivity Fingerprints, ECFP) 。
这项结果表明,在许多化学问题中,高质量的数据和严谨的数据处理,其重要性超过了算法本身的复杂性。
可解释的预测
该研究的价值不止于预测精度,更在于打开了模型的「黑箱」。它结合了两种方法:
首先是保形预测 (Conformal Prediction) 。这种方法让模型在给出预测的同时,量化自身的不确定性。它不再仅仅判断「分子有选择性」,而是提供一个置信度,例如「该分子具备选择性的置信度为 95%」。这种包含不确定性评估的预测,为决策提供了依据。
其次是反事实分析 (Counterfactual Analysis) 。该方法让 AI 能够用化学的逻辑进行解释。模型可以指出:「这个分子的高选择性,关键在于其尾部的磺酰胺基团。如果将其替换为羧酸,选择性便会下降。」
这种分析将模糊的预测转化为清晰、可验证的科学假说,直接指导后续的分子设计。
研究团队将整个分析流程整合成一个名为CAInsight的图形界面软件,使其从学术研究成果转化为可供实际项目使用的工具。
这项工作将「可解释 AI」成功应用于药物发现。它表明,AI 的角色并非取代研究者的「神谕」,而是一个能够与之对话、理性评估自身局限的「高级顾问」。
📜Title: Interpretable Machine Learning Unveils Carbonic Anhydrase Inhibition via Conformal and Counterfactual Prediction
📜Paper: https://doi.org/10.26434/chemrxiv-2025-m69tw
3. 零样本分子生成:SiMGen 的小数据魔法
AI 制药领域普遍认为模型越大、数据越多,效果越好。模型参数动辄上亿,训练数据常覆盖整个 ZINC 数据库。一篇发表于《自然 - 通讯》的文章则提出了另一种思路。
SiMGen 的工作方式不同。它不依赖暴力学习,而是利用「相似性」原理。好比一位顶尖的米其林大厨,无需学习成千上万份菜单(海量数据),只需品尝几道招牌菜(小型参考分子集),便能领悟每种风味(局部原子环境)的精髓。之后,他就能融会贯通,创造出全新的、甚至更复杂的菜肴。
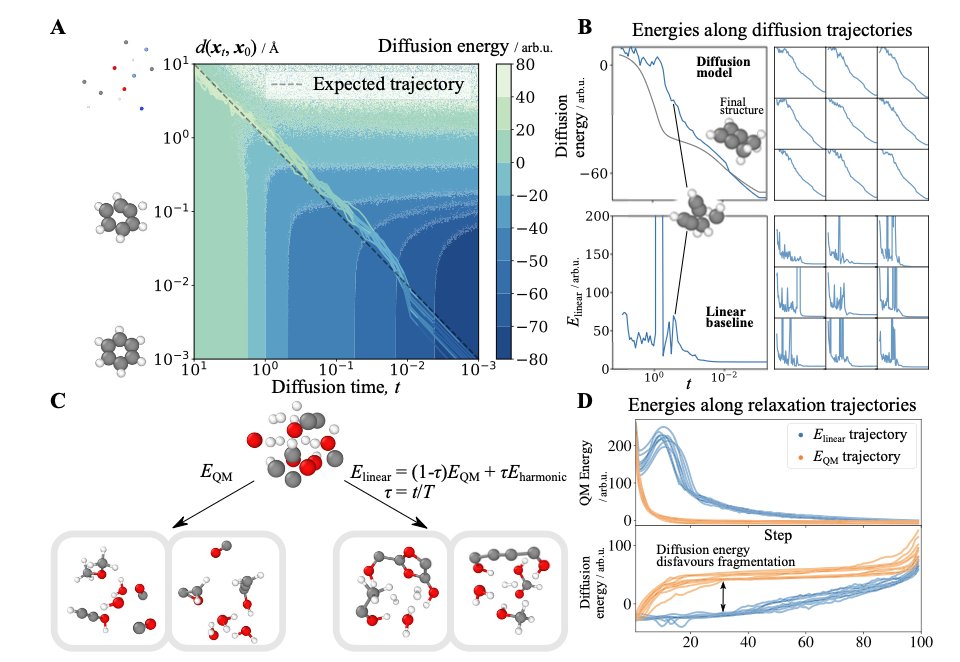
该方法的核心在于「局部性」。传统扩散模型生成大分子,如同一次性雕刻整块巨石,容易在细节处出错。SiMGen 逐个构建原子环境,好比用小块积木搭建复杂城堡。因此,论文中提到它能生成超过 100 个重原子的分子,也合乎情理。这对研究大环或天然产物的学者是一大助益。
它处理元素类型的方式也很有特点。模型没有训练复杂的「炼金术」网络来预测原子替换,而是采用了一种改进的粒子群优化(Particle Swarm Optimization, PSO)算法。该算法的本质是一种智能试错:「将此处的碳原子换成氮原子,结果是否更接近参考分子?如果是,就保留更改。」这种方法简单、直接且有效,避免了大量计算。
对药物研发人员而言,片段引导功能价值很高。研究者常有一个特定的化学片段,例如某个弹头或一个能与靶点紧密结合的苯环,希望 AI 能将其嵌入并补全分子剩余部分。SiMGen 能够实现这一点。它将研究者的想法(空间限制或化学片段)转化为生成过程中的一种约束,引导分子结构向期望的方向发展。这使 AI 从一个不可控的创造者,转变为可协作的设计师。
SiMGen 挑战了对分子生成模型的固有认知,证明了优雅的算法设计和化学直觉,其价值有时会超过海量数据与算力。它为大型模型提供了一种补充,是一种更贴近化学家思维的新工具。模型的局限在于,参考集的质量决定了生成结果的上限。
📜Title: Zero-shot 3D molecular generation via similarity kernels
📜Paper: https://www.nature.com/articles/s41467-024-50963-3
💻Code: https://github.com/RokasEl/simgen