
目录
- 这款叫 DeepADR 的新模型,用上了新潮的 KAN 网络,把药物结构、靶点和副作用的语义信息捏合在一起,想在药物研发的早期就揪出那些潜在的“雷”。
- 在多组学研究中,用 SHAP 来解释深度学习模型就像掷骰子——结果随机性太强,想靠它精准锁定关键生物分子,得加倍小心。
- GenoMAS 利用一个异构的 AI 智能体(Agent)团队,成功实现了对复杂基因表达数据分析的自动化,其表现超越了现有技术,为生物信息学分析带来了突破性的潜力。
1. DeepADR:用 KAN 网络预警药物“翻车”风险
预测药物不良反应(ADR),在药物研发中一直是一项令人头痛的挑战。它犹如幽灵般,常常在你最不经意之时显现,轻易地摧毁一个原本前途光明的候选分子,甚至将已上市的重磅药物拉下神坛。每年有多少项目因此遭遇失败,真是数不胜数。
现今,一篇新的文章提出了 DeepADR 模型,宣称要为这一长久以来的难题提供新解。
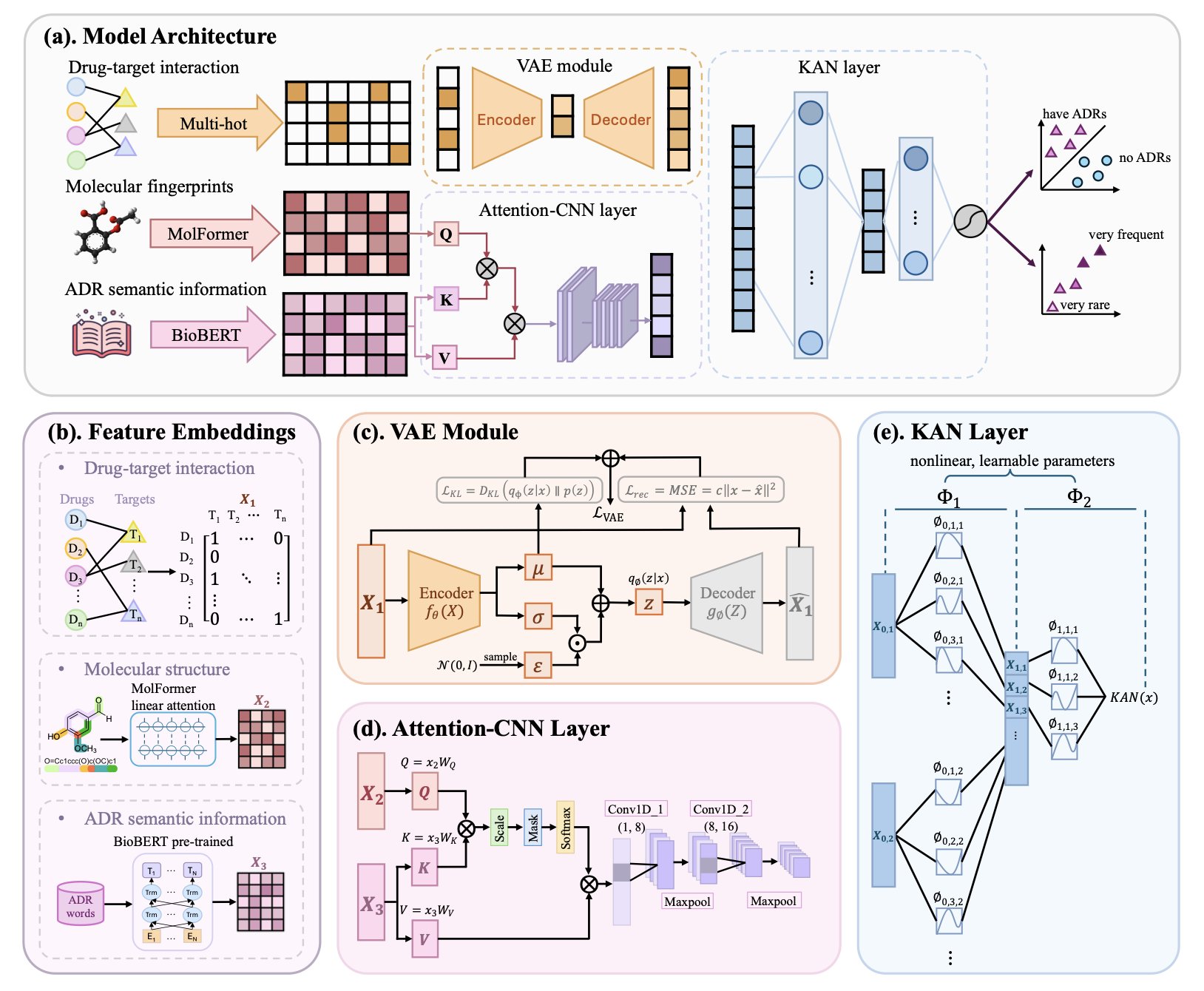
研究者们并未依赖复杂的数据,而是使用了我们项目组手中最常见的三种信息:分子的二维结构图、已知的生物靶点,以及从 SIDER 等数据库中提取的副作用文本描述。将这三者有机结合,思路便清晰明了。一个分子的化学结构决定了其与哪些蛋白(靶点)之间的相互作用,而这些生物学事件最终将表现为某种生理或病理现象(即副作用)。这一逻辑链条十分明确。
真正的亮点,是他们用的算法。
他们没有用 Transformer 或者什么图神经网络,而是搬出了最近有点火的 Kolmogorov–Arnold Network,简称 KAN。
这是个很有意思的选择。
传统的深度学习模型,很多时候就像个密不透风的黑箱。你把数据扔进去,它吐出个结果,至于里头发生了什么,天知道。但 KAN 不太一样。你可以把它想象成一个由无数个微型、可解释的数学公式组成的灵活网络,而不是一个庞大、僵化的计算怪兽。这就让它在处理那些纠缠不清的、乱麻一样的非线性关系时,比那些靠“大力出奇迹”的模型更有章法。
药物毒性恰恰就是最典型的非线性问题。
一个分子可能蹭到十几个靶点(脱靶),每个靶点都可能引发一连串的下游生物学反应,这些反应之间又会相互叠加、放大或者抵消,最后才表现为一个特定的副作用。想用简单的线性模型去描绘这幅景象,简直是天方夜谭。而 KAN 这种结构,理论上讲,就是为了梳理这种复杂关系而生的。
结果怎么样?
看起来还真不错。论文里说,DeepADR 不仅在预测“有没有”副作用这件事上比现有方法更准,还能预测副作用的“发生频率”。这一点非常关键。对于医生和监管机构来说,一个发生率 50% 的轻微头痛和一个发生率 0.1% 的致命心血管事件,完全是两个概念。
对罕见副作用的敏感度。这可是要了命的难题。那些千分之一、万分之一概率的毒性,在几百人的临床试验里几乎不可能被发现,但一旦药物推向市场,面对数百万患者,就可能变成一个随时会引爆的炸弹。如果 AI 能在这方面提供哪怕一点点早期预警,那价值就无法估量了。
当然,话也别说得太满。
这终究是一个回顾性的研究,用的是已有的公开数据。真实世界里,我们每天面对的都是全新的、从未见过的化学结构,以及大量潜在的、未知的脱靶效应。模型在这些新化学实体(NCE)上的泛化能力到底有多强,还得在实际项目中真刀真枪地检验。
而且,这类模型的命门永远是数据质量。公共数据库里的数据质量参差不齐,标注错误、信息缺失都是家常便饭。“垃圾进,垃圾出”,再漂亮的算法也无力回天。
但不管怎么说,DeepADR 把 KAN 这个新工具用在了刀刃上,并且试图解决一个药物研发里最头疼的问题之一。这至少给我们提供了一个新视角,一个可能在未来帮我们少踩点坑的新工具。
📜Title: DeepADR: Multi-modal Prediction of Adverse Drug Reaction Frequency by Integrating Early-Stage Drug Discovery Information via Kolmogorov-Arnold Network…
📜Paper: https://www.biorxiv.org/content/10.1101/2025.07.29.667409v1
💻Code: https://github.com/Wjt777/DeepADR
2. 别太信 SHAP!深度学习模型解释性可能只是幻觉
SHAP (Shapley Additive Explanations) 声称能够揭示深度学习的“黑箱”,告知我们模型究竟重视哪些特征。听起来确实令人向往?然而,最近一篇论文却泼了我们一盆冷水,指出在多组学数据分析这一复杂场景中,SHAP 可能比你想象的更加“善变”。
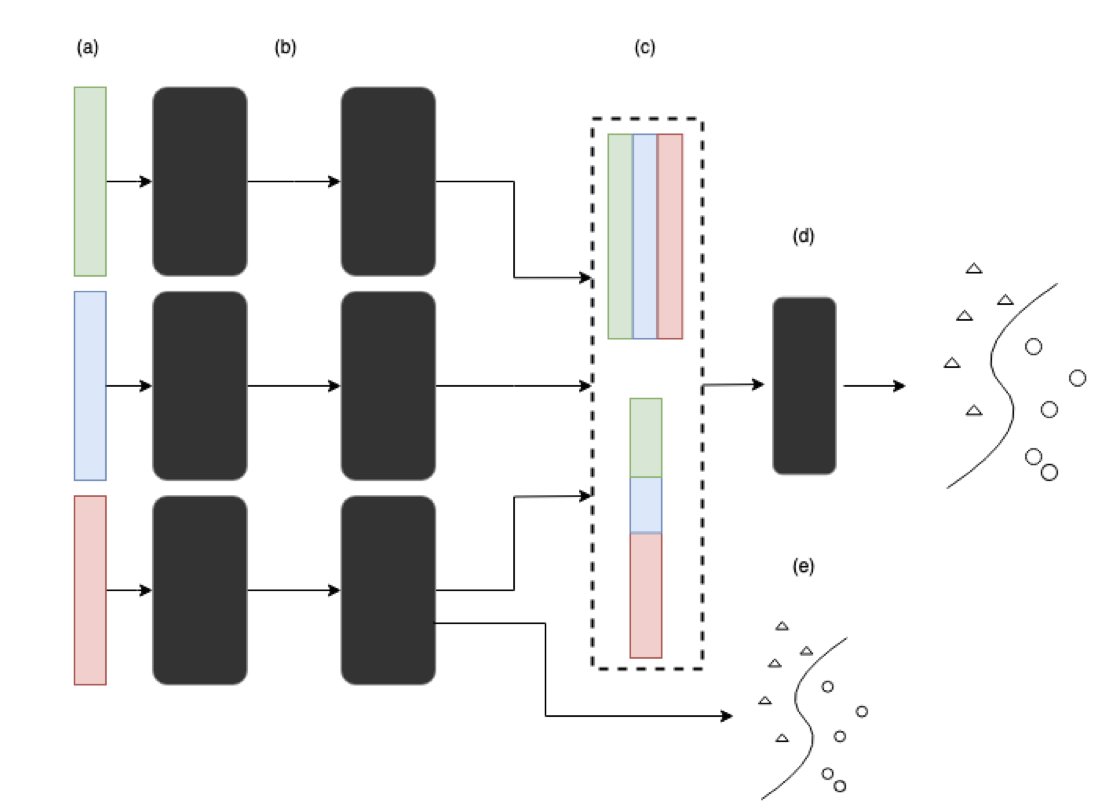
想象一下,你训练了两个一模一样的模型,用的是完全一样的数据,仅仅因为初始化的随机种子不同,最后 SHAP 给出的最重要的 10 个基因(或蛋白质)就可能面目全非。这还怎么玩?做药物发现,最想知道的就是那个“金钥匙”靶点,结果 AI 给了你一串随机号码,这不就是开了个天大的玩笑吗?
研究者们直接在两个真实的多组学数据集上动了手。他们构建了深度学习模型,然后像做实验一样,反复训练。结果呢?特征重要性的排名波动大得惊人。今天这个基因是 Top 1,明天可能就掉到 50 名开外了。他们还往数据里掺“噪音”,模拟真实世界里不那么干净的数据,发现 SHAP 的排名被搅得更乱了。试图通过动态调整网络层的大小来“稳住”SHAP,效果也并不理想。
虽然单个特征的排名像坐过山车,但如果你把 SHAP 选出来的排名前 N 的特征打包,扔给一个随机森林模型去做分类或聚类,性能居然还挺稳定。
这说明什么?
可能 SHAP 选出的特征“整体”上确实抓到了一些信号,但具体到哪个特征贡献最大,它自己也说不清楚。这就像一个侦探团队,总能破案,但你问他们谁是头号功臣,他们每次给出的答案都不一样。对于需要精准打击单一靶点的药物研发来说,这种“集体主义”的正确性帮助有限。
下次当你的 AI 团队兴高采烈地拿着一张 SHAP 图,告诉你他们找到了驱动疾病的关键基因时,你最好先冷静一下。问问他们:“这个结果跑了多少次?每次的结果一致吗?”这篇论文给我们的血泪教训是,至少要跑个十次八次,取个平均值,看看结果的方差有多大。如果某个特征在每次运行中都稳居前列,那它可能才是真正的“金子”。
归根结底,AI 解释性工具远未到可以盲目信任的阶段。保持怀疑,反复验证,这才是科学家的基本素养,在 AI 时代尤其如此。
📜Title: Consistency of Feature Attribution in Deep Learning Architectures for Multi-Omics
📜Paper: https://arxiv.org/abs/2507.22877
💻Code:
3. AI 智能体团队 GenoMAS:自动化基因分析新范式
处理基因表达数据有时就如同在龙卷风中给猫梳毛,既混乱又难以把握要点。每一个接触过基因组学的人,都能切身体会到被庞大数据和复杂流程所支配的恐惧。
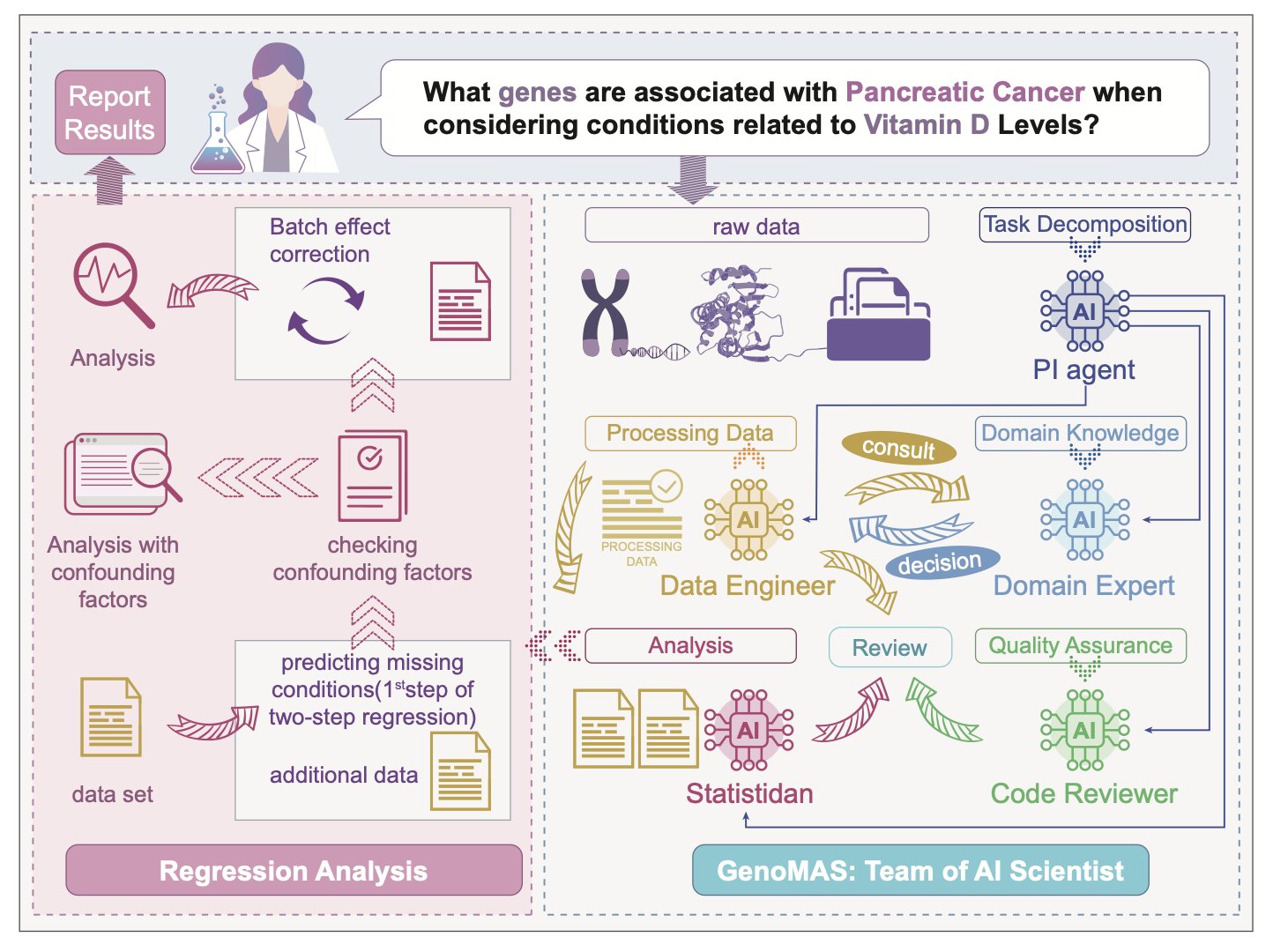
研究人员试图建立一个全面的AI 智能体团队来解决这一难题。可以将其视为一个顶尖的药物研发小组。你不会希望由单一人员承担所有任务,对不对?你需要生物信息学专家、统计学家、数据工程师……GenoMAS 正是为了实现这一目标,只是通过大语言模型(LLM)来组建团队。
它配置了一个由六名成员组成的“AI 小组”,每位成员各有专长——有的擅长编程,有的擅长逻辑推理,还有的负责审查和验证。它们在一个共享的“数字实验台”上协同合作。
论文里有个很有意思的发现:这个由不同专家组成的异构团队,干起活来比让一群能力相同的“通用型”AI 一起上,效率要高得多。这其实一点也不奇怪,顶尖的科学研究就是这么干出来的。
但真正的魔法在于这个团队的管理方式。
系统给它们一个高层级的“作战计划”,就像你在白板上勾勒的实验大纲,但它绝不搞微观管理。这些 AI 智能体可以即兴发挥,自己编写代码来解决眼下的问题。它们还会从错误中学习。如果一段代码跑得通、效果好,就会被存进一个共享的“工具箱”里供日后使用;如果失败了,就换条路走。这种自我纠错和迭代的能力至关重要,它让系统从一个脆弱的“脚本执行器”变成了一个足智多谋的“博士后”。
我们来看看结果。
在基因识别任务上,它的 F1 分数比之前的 SOTA 高出 16.85%,这绝对是质的飞跃。这意味着它找到的基因 - 表型关联不仅仅是统计上的巧合,而是很可能具有真实的生物学意义。研究者们在 GenoTEX 这个业内公认的、相当棘手的基准测试平台上验证了它,GenoMAS 在从数据集筛选到最终统计分析的整个工作流中都拔得头筹。这表明它不是一个只能在理想环境下运行的玩具,而是有潜力处理端到端真实科研任务的系统。
我们可能正在见证一个转变:AI 不再仅仅是个“工具”,而正在成为“合作者”。对于那些没有专门生信团队的小型实验室或初创公司,GenoMAS 这样的系统简直是天赐之物,它有望让顶级的基因组分析能力变得大众化。
当然,它在处理那些真正混乱、充满噪音的真实世界数据时表现如何?它能否对全新的生物学通路进行推理?
这些是它接下来必须回答的问题。但就目前而言,GenoMAS 无疑是朝着正确方向迈出的、令人兴奋的一大步。
📜Title: GenoMAS: A Multi-Agent Framework for Scientific Discovery via Code-Driven Gene Expression Analysis
📜Paper: https://arxiv.org/abs/2507.21035