
目录
- 这个 AI 不仅预测分子活性,它还被训练来解释我们最头疼的「活性悬崖」现象,这让它终于有点像个能沟通的同事了。
- 通过先让 AI 学习「通用化学知识」,再在少量抗菌数据上「专科训练」,这个迁移学习框架成功地在超十亿分子库中,淘出了亚微摩尔级别的抗菌新药。
- 这个 AI 框架通过 GFlowNets 生成罕见的药物相互作用(DDI)样本,来「平衡」训练数据,从而让预测模型不再只认识「大路货」。
1. AI 终于学会解释化学里最头疼的「悬崖」问题
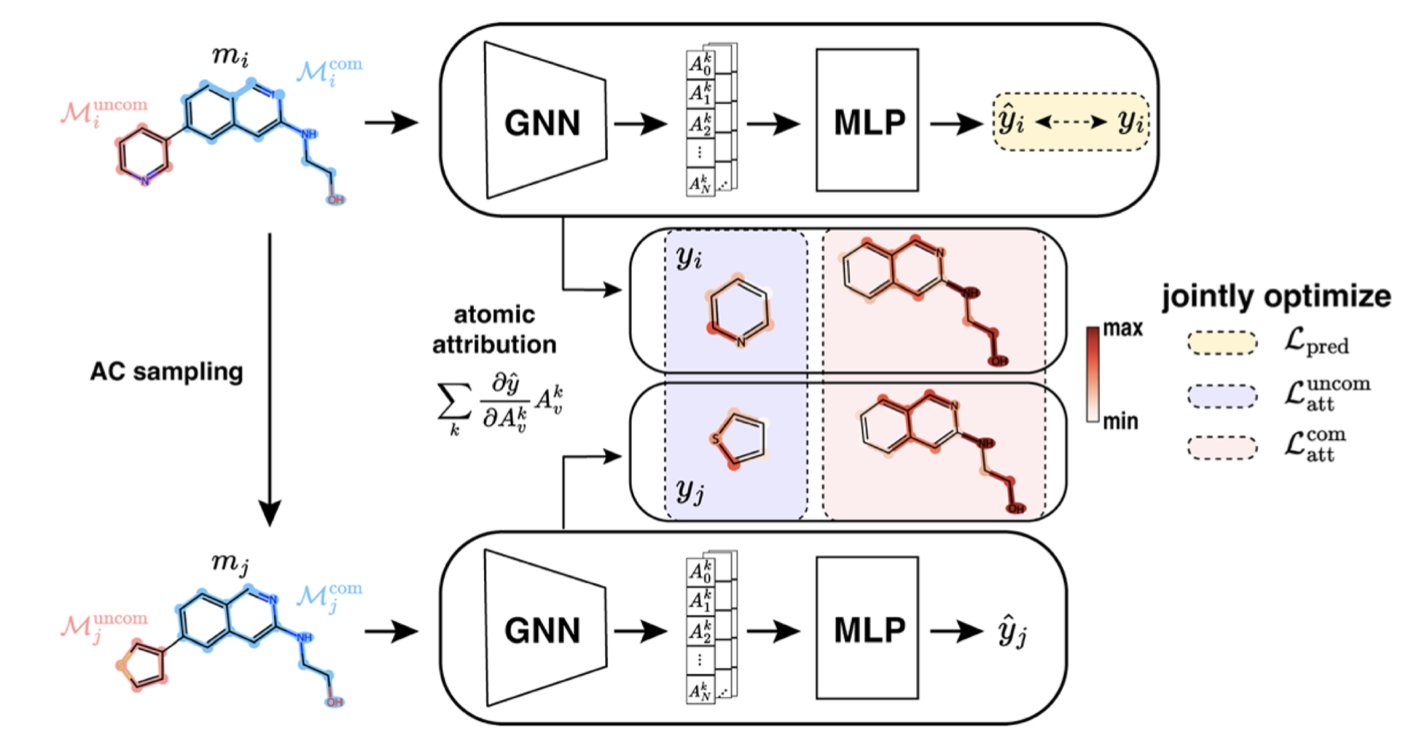
直接攻击药物化学中的「活性悬崖」——两个分子长得像双胞胎,活性却差了十万八千里——这是传统 AI 模型的噩梦。
你辛辛苦苦设计合成了一个分子,活性不错,10 纳摩尔。然后你信心满满地想:「如果我把这个甲基换成乙基,多一个碳,疏水性更好,活性肯定能翻倍!」结果呢?你花了两周时间做出新分子,一测活性,10 微摩尔,直接掉了 1000 倍。你的笑容凝固在脸上,完美的假设和理论,瞬间摔得粉碎。
而我们现有的那些 AI 模型,在面对这种「悬崖」时,基本都废了。它们要么直接无视,要么给出一个错得离谱的预测,然后耸耸肩,给你留下一个无法解释的黑箱。一个不能告诉你「为什么」的 AI,在药物发现中,价值极其有限。我们需要的不是一个只会报数的算命先生,我们需要的是一个能和我们讨论、能告诉我们它思考过程的同事。
ACES-GNN 试图去理解并解释「活性悬崖」的 AI。
它在训练的时候,不只是给 AI 一堆分子和它们的活性数据,让它自己去「悟」。它还额外给了一份「标准答案」,这份答案就是化学家对这些「悬崖」的解释。比如,对于一对活性悬崖分子,化学家会指出:「你看,这两个分子唯一的区别,就是这个氯原子。很可能就是这个氯原子,和蛋白口袋里的某个氨基酸形成了关键的相互作用,才导致了活性的巨大差异。」
ACES-GNN 就被强迫去学习这种「解释」。它的损失函数里,有一项专门惩罚那些「解释」得不对的预测。如果 AI 把注意力放在了分子中某个无关紧要的尾巴上,而不是那个关键的氯原子,它就会被「扣分」。这就像一个严格的老师,不只要求学生给出正确答案,还必须写出正确的、符合逻辑的解题步骤。
这么做的结果是什么?
首先,这个 AI 生成的「归因图」——也就是它认为分子中哪些部分对活性贡献最大——变得和化学家的直觉高度一致。它真的开始「说人话」了。
当一个 AI 被训练得能更好地「解释」问题时,它「预测」问题的能力也跟着变强了。这篇论文的数据显示,ACES-GNN 在 30 个不同的药物靶点上,预测准确性和归因质量都稳定地优于那些没有经过「解释训练」的普通 GNN 模型。这说明,强迫 AI 去理解底层的因果关系,而不仅仅是学习表面的相关性,能让它变得更强大、更鲁棒。
我们是不是马上就能拥有一个全知的 AI 化学家了?想得美。
这个方法依赖于我们能提供高质量的、关于活性悬崖的「人类解释」作为训练数据,而这本身就不是一件容易的事。而且,它仍然是一个模型,它仍然会犯错。
📜Title: ACES-GNN: Can Graph Neural Network Learn to Explain Activity Cliffs?
📜Paper: https://pubs.rsc.org/en/content/articlelanding/2025/dd/d5dd00012b
💻Code: https://github.com/Liu-group/XACs
2. AI「借脑」筛药,在十亿分子中发现新抗生素
![]()
寻找新的抗生素,是我们这个时代最紧迫的医学挑战之一。
那些曾经的「超级细菌」(ESKAPE 病原体),正变得越来越耐药,而我们的新药研发管线,却干涸得像撒哈拉沙漠。一个主要的原因是,传统的筛选方法,就像是在大海捞针,效率极低。
AI 虚拟筛选给了我们新的希望。理论上,我们可以在电脑里,快速评估数以十亿计的分子,找出那些最有潜力的候选者,再去实验室里验证。但这又带来一个新问题:训练一个好的 AI 预测模型,需要大量高质量的、已知的抗菌活性数据。而这类数据,恰恰是非常稀缺和宝贵的。
这就成了一个「先有鸡还是先有蛋」的悖论:为了找到新药,你需要一个好模型;为了训练一个好模型,你需要大量已知药物的数据。
这篇来自 bioRxiv 的新论文,就为我们展示了一种打破这个悖论的方法,它的名字叫做「迁移学习」(transfer learning)。
这个概念,其实在我们的日常学习中很常见。一个学好了物理的学生,再去学化学,通常会比一个零基础的学生快得多。因为他已经掌握了很多底层的、可迁移的知识,比如原子、能量、相互作用等。他不需要从头学起。
这个研究团队对他们的深度图神经网络(DGNN)模型,做的就是类似的事情。
- 第一步:通识教育(Pre-training)。
他们没有一开始就把模型扔到那个数据稀缺的「抗菌专业」里去。他们先找来了一些数据量巨大,但与抗菌活性完全无关的「通识教材」,比如:- 大量分子的对接分数(docking scores):这教会模型,什么样的分子形状,更容易「塞」进蛋白质的口袋里。
- 大量分子的结合亲和力(binding affinities):这教会模型,什么样的化学基团,更容易和蛋白质产生相互作用。
在这个阶段,模型学习到的,不是「如何杀死细菌」,而是更底层的、更通用的「化学语言」和「分子相互作用的物理规律」。它成了一个博学的「化学通才」。
- 大量分子的对接分数(docking scores):这教会模型,什么样的分子形状,更容易「塞」进蛋白质的口袋里。
- 第二步:专业课集训(Fine-tuning)。
当模型完成了「通识教育」后,他们才把它送到那个数据量虽小,但专业性极强的「抗菌专业」里去深造。他们用那些来之不易的、真实的抗菌筛选数据,对这个已经很「博学」的模型进行微调。
因为模型已经有了坚实的化学基础,所以它不再需要成千上万个样本,才能学会识别抗菌分子的特征。它只需要几十、几百个例子,就能迅速地将它的通用知识,「迁移」和「适配」到这个特定的新任务上来。
这个「先通识,后专业」的策略,效果如何?
他们用这个训练好的模型,去「检阅」了 ChemDiv 和 Enamine 这两个商业化合物库中,超过十亿个虚拟分子。模型从中挑选出了它认为最有希望的 156 个候选者。
然后,激动人心的时刻到来了:他们真的去实验室里,合成了或者购买了这些分子,并在大肠杆菌上进行了测试。结果,命中率高达 54%!这意味着,模型挑出来的化合物里,超过一半都真的有抗菌活性。这在传统的药物筛选中,是难以想象的。
更重要的是,这些被发现的化合物中,有很多都表现出了亚微摩尔级别(sub-micromolar)的强效活性,而且其中一些还具有广谱抗菌效果。从化学结构上看,它们也包含了一些新颖的骨架,这意味着 AI 成功地带领我们,航行到了未曾探索过的「化学宇宙」的新区域。
这我们如何在一个「数据贫瘠」的领域,利用 AI 进行药物发现,提供了一个强有力的范例。有时候,最有效的方法,不是在一个小池塘里反复打捞,而是先让你的 AI 学会「游泳」的通用技能,然后再把它扔进任何一个你感兴趣的池塘里。
📜Title: Transfer Learning Enables Discovery of Submicromolar Antibacterials for ESKAPE Pathogens from Ultra-Large Chemical Spaces
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.07.669197v1
3. AI 左右互搏,专攻罕见药物相互作用
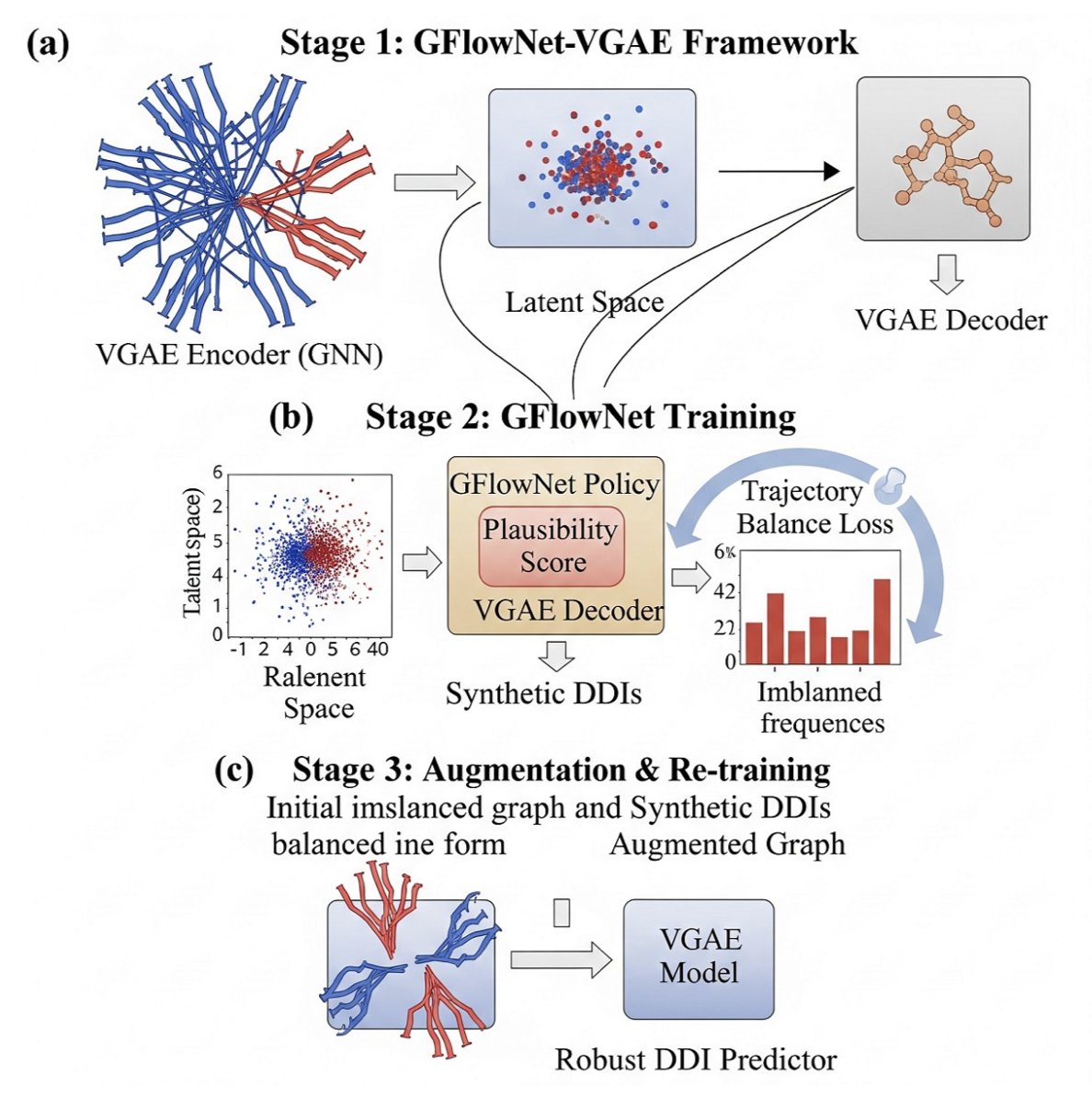
在临床药理学中,药物 - 药物相互作用(DDI)是个永恒的难题。当一个病人同时服用多种药物时(这在老年患者中非常普遍),这些药物在体内可能会相互「打架」,导致药效降低,甚至产生严重的毒副作用。能够准确预测哪些药物组合是「危险品」,对用药安全至关重要。
我们当然想用 AI 来干这件事。但这里有一个障碍,叫做 「类别不平衡」(class imbalance) 。
想象一下,你要训练一个 AI 来识别动物。你给它看了一百万张猫的照片,十万张狗的照片,但只给它看了一张水豚的照片。结果可想而知,这个 AI 会变成一个顶级的「猫咪识别器」,一个还不错的「狗狗识别器」,但它很可能会把任何长得有点像啮齿动物的东西都当成猫,因为它见过的水豚实在太少了。
DDI 数据集就是这个样子。某些类型的相互作用,比如通过 CYP3A4 酶代谢的药物,数据多如牛毛。而另一些罕见但可能致命的相互作用,比如某些药物对转运蛋白的抑制,相关的数据可能就那么几条。用这种偏科严重的数据训练出来的模型,自然也就成了「偏科生」,对那些罕见但重要的风险「视而不见」。
这篇来自 arXiv 的论文,就提出了一个很聪明的解决方案,来纠正模型的「偏科」问题。他们的核心思想,不是去现实世界里辛辛苦苦地找更多的「水豚」照片,而是用 AI 自己,去画出成千上万张逼真的「假水豚」照片。
他们用的「画家」,是一种叫做Generative Flow Networks (GFlowNets)的生成模型。
整个过程可以分成三步,像一场精心编排的「左右互搏」:
第一阶段:初学乍练。
他们先用一个变分图自动编码器(VGAE),在现有的、不平衡的 DDI 数据上进行预训练。VGAE 的作用,是学习如何将每个药物分子,编码成一个浓缩了其化学特征的「数字指纹」(embedding)。这个阶段,模型学会了对药物的基本「相面」之术,但因为它看的数据是偏的,所以它对罕见相互作用的理解还很肤浅。第二阶段:创造性练习。
这是最关键的一步。他们把第一阶段学到的「数字指纹」,交给了 GFlowNets 这个「画家」。GFlowNets 的任务,就是生成新的、合成的 DDI 样本。但它不是瞎画,而是有一个非常明确的目标,这个目标由一个巧妙的「奖励函数」来定义。
这个奖励函数的设计是:你生成的 DDI 样本,所属的类别越罕见,你得到的奖励就越高。这就像是告诉画家:「别再画猫了,画一只水豚,我给你双倍的钱!」
这个机制,驱使着 GFlowNets 不断地去探索和生成那些在原始数据集中数量稀少的 DDI 类型的样本。它不仅要保证生成的样本看起来「合理」(plausible),还要保证它们是「稀有品」。第三阶段:融会贯通。
最后,他们把 GFlowNets 生成的这些高质量的「假水豚」照片,和原始数据集中所有的「真猫」、「真狗」、「真水豚」照片混合在一起,形成了一个数据更平衡、更多样化的增强数据集。然后,他们用这个新的、更丰富的数据集,去重新训练第一阶段的那个 VGAE 模型。
经过这番「补课」,VGAE 模型就不再是那个只认识猫的「偏科生」了。它对那些罕见的相互作用类型,有了更深刻的理解,预测性能自然也就水涨船高。
实验结果也证明了这一点。经过 GFlowNets 增强后,数据集的多样性指标(如香农熵)显著提升,最终的 DDI 预测模型在所有相互作用类型上,特别是那些罕见类型上,都表现出了更好的性能。
无论是预测罕见病、基因组变异,还是药物不良事件,这种「用 AI 创造数据来教 AI」的思路,都可能成为我们未来工具箱里的一件利器。
📜Title: GFlowNets for Learning Better Drug-Drug Interaction Representations
📜Paper: https://arxiv.org/abs/2508.06576