
目录
- 这个 AI 模型通过「正向引导」和「负向引导」,像调酒师一样,一边加入想要的成分,一边减少不好的杂味,来设计更精准、更安全的药物分子。
- 通过学习 MRI 影像中哪些「患者特征」对抗癫痫药物有反应,这个 AI 模型甚至能预测那些它从未见过的「新药」的治疗效果。
- 尽管 AI 炒作不断,这项大规模基准测试表明,简单的老式化学指纹法在分子表征上,仍然优于几乎所有新的深度学习模型。
1. ActivityDiff:AI 像调酒师一样,精准调控药物活性
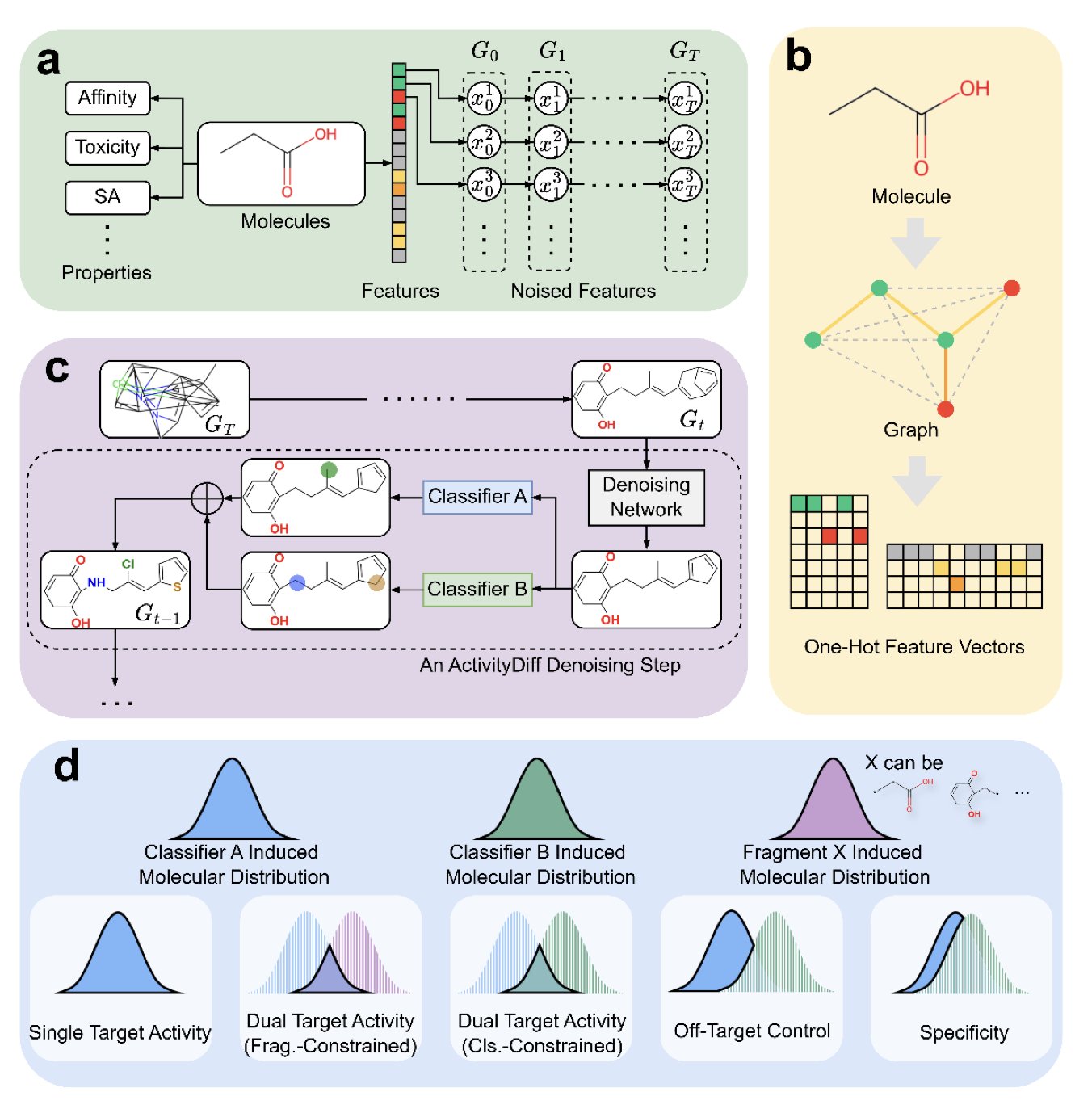
我们想让一个分子对我们感兴趣的靶点(比如某个致病蛋白)有很强的活性,y 也就是「药效」;同时,我们又希望它别去招惹那些我们不想碰的靶点,因为那通常意味着「副作用」或「毒性」。
过去,我们做这件事有点像「打地鼠」。你优化一个分子,增强了对 A 靶点的活性,结果它对 B 靶点的活性也意外地增强了,带来了不想要的副作用。然后你又去修饰分子,试图降低对 B 的活性,结果可能又影响了对 A 的活性。这个过程非常繁琐,而且充满了妥协。
现在,扩散模型(diffusion model),给这件事带来了新思路。我们可以把扩散模型想象成一个技艺高超的雕塑家。你先给他一堆随机的「黏土」(一堆随机的原子),然后告诉他:「我想要一个看起来像大卫的雕像」。他就会一步一步地,把这堆随机的黏土塑造成你想要的样子。
ActivityDiff 就是在这个「雕塑」过程中,给了雕塑家更精细的指令。它不只是说「我想要个大卫」,它还说「我想要个大卫,但他绝对不能看起来像个掷铁饼者」。
它是怎么做到的呢?通过所谓的 「正向引导」 和 「负向引导」 。
这背后其实是几个独立的、小型的 AI 模型(药物 - 靶点分类器)在协同工作。你可以把它们想象成几个专业的「艺术评论家」。
这样一来,整个分子生成过程,就像是在一个多维空间里寻找一个最优点。正向引导把它往「高药效」的山峰上拉,负向引导把它从「高毒性」的悬崖边推开。
这个方法最妙的地方在于它的模块化。那个负责雕刻的「雕塑家」(扩散模型本身)可以保持不变。如果你今天想设计一个同时激活 A 和 C 靶点,但避开 D 靶点的分子,你不需要重新训练那个庞大的雕塑家。你只需要训练几个新的、小巧的「艺术评论家」(针对 A、C、D 靶点的分类器),然后把它们组合起来,去指导同一个雕塑家工作就行了。这大大降低了计算成本和时间。
作者们用实验证明了这套方法的有效性。他们不仅成功生成了高活性的单靶点和双靶点分子,还完成了一些更复杂的任务,比如在一个已有的分子片段上「长出」新的结构,同时保证新长出来的部分满足特定的活性要求。这就像是告诉你:「我这里有个雕塑的底座,你帮我在上面雕个大卫的头出来,但要保证他看起来不会去掷铁饼」。
当然,分类器的准确性直接决定了引导的质量。如果你的「艺术评论家」自己眼神不好,那他也给不出什么好建议。还有,化学空间的广阔性意味着,AI 生成的全新分子,其真实活性最终还是要靠湿实验(wet lab)来验证。
📜Title: ActivityDiff: A diffusion model with Positive and Negative Activity Guidance for De Novo Drug Design
📜Paper: https://arxiv.org/abs/2508.06364
2. AI 读脑 CT,帮你预测哪种癫痫药最有效
在神经内科癫痫治疗领域,有一个让医生和患者都头疼不已的现实:选择抗癫痫药物(ASMs)很大程度上是一场「试错」之旅。
我们有几十种药物,但没有一个可靠的方法能在用药前,就知道哪一种对特定患者最有效。于是,治疗过程就变成了:试一种药,观察几个月,没效果或者副作用太大,再换下一种。这个过程对患者来说,是身体和心理上的双重折磨。
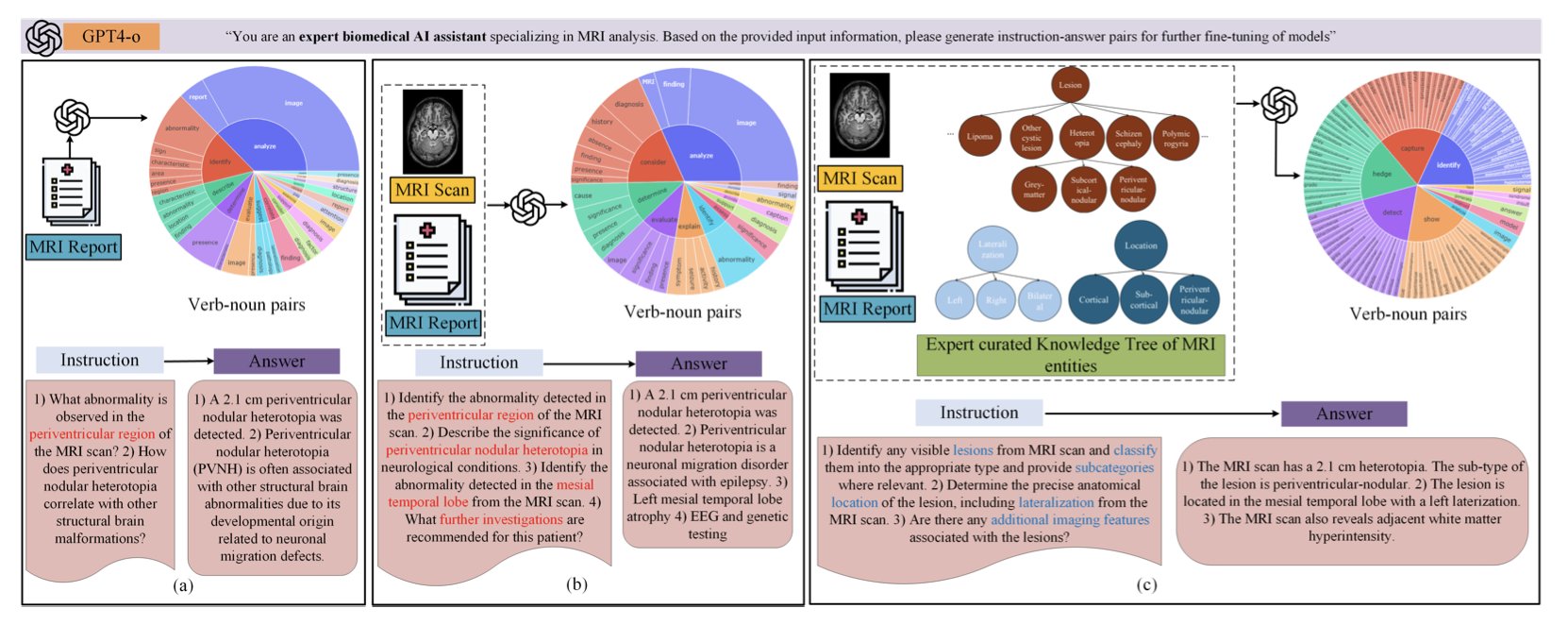
所以,任何能减少这种「试错」成本的方法,都值得我们认真看一看。这篇预印本论文就提出了一个很有意思的尝试:他们想用 AI,只通过患者的 MRI 脑部扫描和放射科医生的报告,来预测不同 ASMs 的治疗效果。
想法本身不新,但他们的做法有几个巧妙之处。
首先,他们用的不是普通的 AI 模型,而是一个生物医学领域的「视觉 - 语言」基础模型。这意味着这个模型在训练之初,就已经「见过」大量的医学影像和相关的文本报告。它天生就对医学术语和影像特征有感觉,而不是像一个通用模型那样,需要从头学起什么是「海马体硬化」。它能同时处理像素(MRI 影像)和文字(报告),并试图将两者联系起来。
其次,也是这个研究最核心的创新点,是一种叫做TREE-TUNE的框架。直接把 MRI 报告扔给 AI 进行微调,效果并不理想,因为报告里信息繁杂,AI 很容易「迷路」,抓住一些无关紧要的关联。TREE-TUNE 就像是给了 AI 一个「专家导航」。研究者先根据神经科学专家的知识,构建了一棵关于癫痫 MRI 表现的「知识树」。这棵树定义了各种病灶、解剖位置和影像学特征之间的层级和逻辑关系。
然后,在微调模型时,他们用这棵知识树来「指导」AI。AI 在阅读报告时,会被引导着去识别和理解这些在知识树中定义好的关键实体。这就像是给了学生一本带有详细注释和重点标记的教科书,而不是让他自己去图书馆里瞎翻。结果显示,用了 TREE-TUNE 之后,预测的准确性确实比常规的微调方法有了显著提升。
真正让人眼前一亮的,是他们关于「泛化到未见药物」的实验。这是这个领域里一个很大的难题。如果一个模型只能预测它训练时见过的药物,那它的临床价值就大打折扣,因为新药总是在不断出现。
这个团队的做法是,只用四种最常见的 ASMs(比如左乙拉西坦、拉莫三嗪等)和患者的 MRI 数据来训练模型。然后,他们用另外三种模型在训练中从未见过的 ASMs 来进行测试。从逻辑上讲,这怎么可能做到呢?
我的理解是,这个模型学习到的,可能不是「MRI 影像 A 对应药物 X 有效」这种简单的映射。它学习到的,可能是一个更深层次的模式,比如「具有某种特定脑部结构或病理特征(由 MRI 和报告共同反映)的患者亚群,对某一类作用机制的药物反应更好」。因为不同的 ASMs 虽然是不同的分子,但它们的作用机制可能是有重叠的(比如都作用于钠离子通道)。所以,模型通过学习已知药物,掌握了这种「患者特征 - 药物机制」的关联,然后把它推广到了具有相似机制的未知药物上。
当然,对于训练时见过的四种药物,模型的平均 AUC 达到了 71.39%,对于那三种未见过的药物,平均 AUC 是 63.03%。说实话,AUC 63% 在临床预测模型里算不上惊艳,它只是比随机猜测(AUC 50%)好一些。
我们离「AI 精准开药」还很远。但是,对于一个它从未见过的药物,能做出比瞎猜有统计学意义上更好的预测,这本身就是一件非常了不起的概念验证(proof-of-concept)。它证明了这条路是可能走得通的。
这篇论文提供了一个框架。它告诉我们,通过将多模态数据(影像 + 文本)和结构化的专家知识(知识树)相结合,AI 或许真的能帮助我们,在这场漫长而痛苦的「试错」游戏中,为患者找到那把正确的「钥匙」。
📜Title: Adapting Biomedical Foundation Models for Predicting Outcomes of Anti Seizure Medications
📜Paper: https://www.medrxiv.org/content/10.1101/2025.08.07.25333198v1
3. AI 分子模型:老派指纹法仍胜过深度学习
在药物发现的世界里,我们一直想找到一个能和计算机有效沟通的「分子语言」。我们怎么向一个只会处理 0 和 1 的机器,描述一个三维的、充满电子云和微妙相互作用的分子,才能让它理解这个分子的「性格」,并预测它在人体内会干些什么?这是一个困扰了化学信息学几十年的核心问题。
最近几年,随着深度学习的浪潮,答案似乎已经显而易见:用庞大的、在数百万甚至数十亿分子上预训练过的神经网络模型。这些模型,就像是饱读了整个化学文献库的「博士生」,理论上,它们应该能比任何老方法都更深刻地理解分子的「语法」和「语义」,从而生成一种更通用、更强大的分子表征(embedding)。
听起来很美,对吧?问题是,这是真的吗?
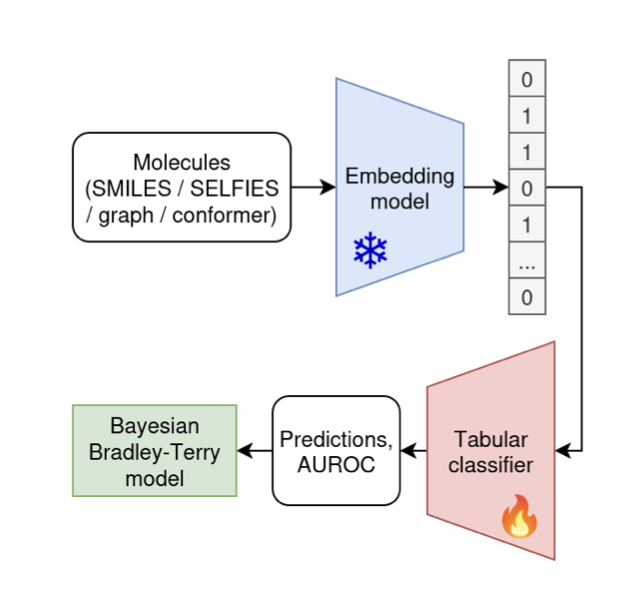
这篇来自 arXiv 的新论文,干了一件「脏活」:他们搞了一场大规模的「笼中对决」。他们找来了 25 个这样的「AI 博士生」(预训练模型),让它们在 25 个不同的「考场」(数据集)里,和一位「老派工匠」(传统的 ECFP 分子指纹法)同场竞技。
ECFP 是什么?你可以把它想象成一种给分子拍「快照」的方法。它不关心分子的深层语法,只是简单粗暴地列出分子中存在的各种局部化学环境和原子片段。这就像描述一个人,不是通过写一篇传记,而是直接列出一串标签:「戴眼镜、有胡子、穿蓝色衬衫、身高 180cm」。这个方法简单、快速,而且已经存在很多年了。
结果出来后,我想很多人可能会感到有些尴尬。
当硝烟散去,那位「老派工匠」ECFP,几乎毫发无损地站在场中央,而周围躺了一地的「AI 博士生」。数据显示,在这 25 个不同的任务中,将近所有花哨的、复杂的神经网络模型,其表现相比简单的 ECFP,「几乎没有或完全没有提升」。
这就像是你花大价钱请来了一位米其林三星大厨,让他给你做个炒鸡蛋,结果发现味道还不如街角早餐店里那个干了三十年的老师傅。
当然,事情总有例外。在这场对决中,有一个名为CLAMP的模型,确实稳定地表现出了比 ECFP 更好的性能。也许通往更好分子表征的道路并没有被完全堵死。但它也反衬出,其他二十多个模型的表现是多么平庸。
那么,这意味着什么?
首先,这是一个 humbling result(令人谦卑的结果)。它提醒我们,在化学信息学这个复杂的领域,不要轻易被模型的复杂性或训练数据的大小所迷惑。一个模型「见过」的分子多,不代表它就真的「理解」了分子。
其次,对于所有在一线做药物发现的计算化学家和数据科学家来说,这是一个非常实用的教训。在你尝试任何一个新的、听起来很酷的分子表征模型之前,请务必,务必先跑一遍 ECFP 作为基准。如果你的新模型连 ECFP 都打不过,那你很可能只是在浪费宝贵的计算资源和时间,去追求一种「看起来很先进」的幻觉。
这篇论文并没有宣判深度学习在分子表征领域的死刑。它像一次精准的「体检」,指出了我们当前的不足。未来的突破,可能不在于构建更庞大、更通用的模型,而在于如何将更多的化学和生物学知识,更巧妙地融入到模型的设计中,就像 CLAMP 可能做到的那样。
📜Title: Benchmarking Pretrained Molecular Embedding Models For Molecular Representation Learning
📜Paper: https://arxiv.org/abs/2508.06199v1