
目录
- Chemsmart 是一个开源的「瑞士军刀」工具包,旨在将计算化学中繁琐的手动流程自动化,让科学家专注于科学思考,而非文件格式转换。
- 这个新框架为 AI 生成模型引入物理学「家教」,通过强化学习奖励化学上稳定、真实的分子,引导 AI 超越简单的形态模仿。
- DrugReasoner 是一款能预测新药获批概率的 AI。它通过对比已知的成功与失败药物来解释预测过程,如同化学家一般推理,从而揭示了预测模型的内部逻辑。
- 结合已知分子片段信息指导并修正传统对接,新流程提升了药物分子结合姿态预测的准确性,展现了数据与物理结合的优势。
- DRUGFLOW 学习「好分子」的完整分布,如同绘制一张完整的藏宝图,由此实现了更真实、可控的分子生成。
1. Chemsmart:告别计算化学的「手工作坊」
亲手操作过量子化学计算的人都清楚:工作的大部分内容是处理繁琐的数据,如同一个数据勤杂工,真正用于思考深奥化学问题的时间很少。
研究者的日常工作流通常如下:用一个软件绘制分子,保存为特定格式;为了计算,再将其转换为另一种格式。接着,需要手动编写输入文件,仔细设定参数,然后提交到计算集群,在等待中频繁检查任务状态。计算结束后,还要从庞大的输出文件中,通过手动或自编脚本提取能量、坐标等关键数据。最后,将这些数据导入可视化软件,生成报告所需的图片。
流程中的每一步都伴随着重复劳动、格式不兼容和人为失误的风险。尽管我们拥有 Gaussian、ORCA 这类强大的计算引擎,却始终在手动为它们「加燃料、清积碳」。
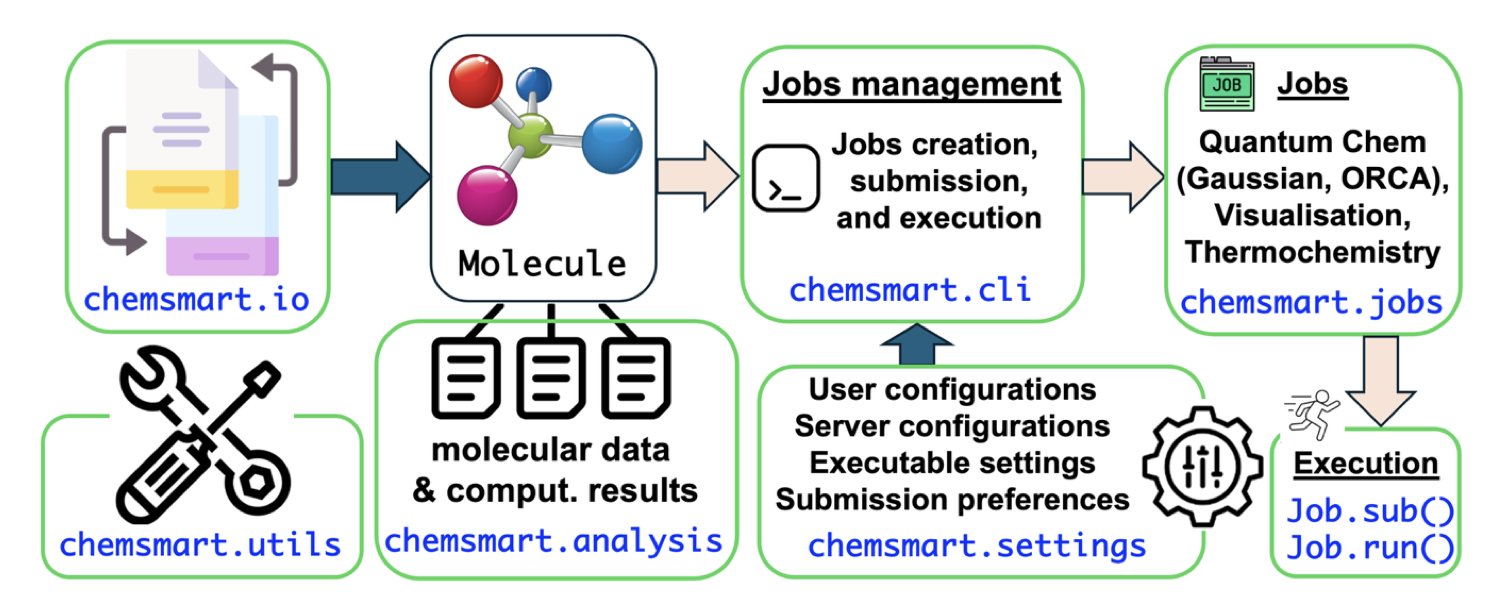
Chemsmart:计算化学的「项目管家」
Chemsmart 工具包的目标是成为管理整个研究流程的「项目管家」,解决研究中的实际痛点。
作为一个开源 Python 工具包,Chemsmart 的设计思路是:将计算化学中最繁琐的手动操作自动化。
它的工作方式如下:
首先,它定义了一个核心的Molecule对象,可以将其理解为一个「通用文件格式」。不同化学软件产生的分子和数据,都能先转换为这个标准对象,从而解决格式不兼容问题,如同为所有软件配备了万能翻译器。
其次,它将完整的计算流程拆分为独立的模块化任务,例如「几何优化」、「过渡态搜索」和「热力学分析」。使用者可以像搭建乐高积木一样,将这些模块组合起来,构建全自动化的工作流。
使用者只需下达指令,例如:「优化这个分子,计算其频率,并生成 PyMOL 可视化文件。」接下来的工作便可完全交给它。Chemsmart 会自动准备输入文件、提交任务、监控进程、解析结果,并将所有文件整理归档。
Chemsmart 带来的改变
研究者得以从重复、缺乏创造性的劳动中解脱,专注于化学问题本身。
对于新手,Chemsmart 降低了入门门槛。使用者无需记忆各种软件晦涩的输入参数,只需调用几个简单的 Python 函数。
对于资深研究者,它提升了工作效率和可重复性。对上百个分子执行标准化的计算流程成为可能,这对构效关系等研究至关重要。
Chemsmart 的所有代码均已开源,并采用模块化设计。因此,它既能作为独立工具使用,也能作为 Python 库,无缝集成到更复杂的定制化分析流程中。
Chemsmart 是一个经验丰富的工程师为解决日常痛点而打造的实用工具箱。对于一线研究者,一个称手的工具箱,其价值有时会超过一个遥远的理论。
📜Title: Chemsmart: Chemistry Simulation and Modeling Automation Toolkit for High-Efficiency Computational Chemistry Workflows
📜Paper: https://arxiv.org/abs/2508.20042v1
2. AI 生成分子:终于学会了物理
与 3D 分子生成模型打过交道的人都清楚一个现实:这些 AI 是顶级的艺术家,却是末流的工程师。
你给它一个任务,它能「画」出一些结构新颖、看起来很美的分子。但把这些「艺术品」放到物理学的放大镜下,问题就来了。键长不对,键角扭曲,原子之间挤得像早高峰的地铁。这些分子在真实世界里根本站不住脚。
我们一直想要一个能懂物理的 AI 工程师。新提出的 RLPF 框架,正是朝这个方向迈出的坚实一步。
给 AI 请个物理家教
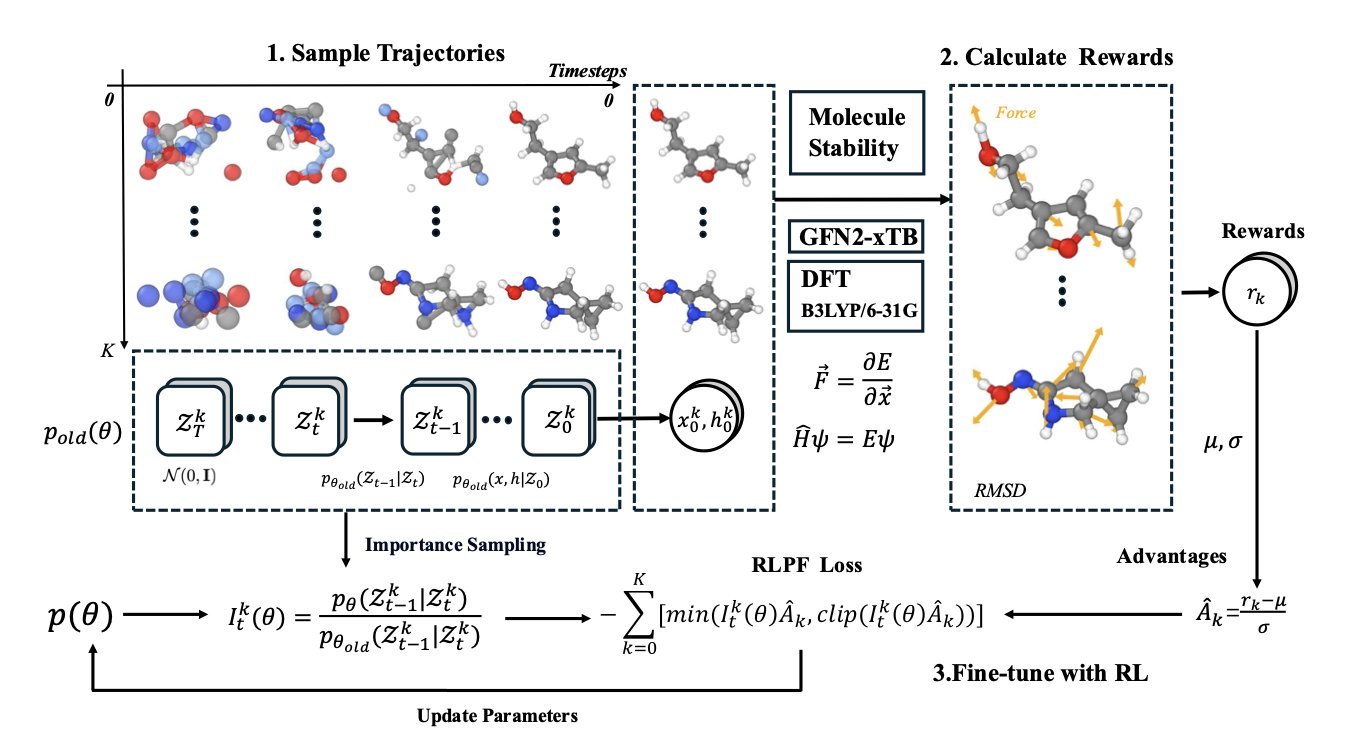
研究者没有去构建一个看过更多数据的、更复杂的 AI。他们另辟蹊径,为 AI 引入了一位物理学「家教」和一套「奖惩机制」。
这个家教是力场,奖惩机制则是强化学习(Reinforcement Learning, RL)。
它的工作方式如下:
扩散模型生成分子的过程并非一步到位。它像雕塑家从一团模糊的「原子云」开始,逐步刻画细节。
RLPF 框架在雕塑家每刻一刀后,都让旁边的物理学「家教」评估一次。
家教(力场)会判断,这一刀是让整个结构的内部应力变大还是变小?是让它更稳定,还是更接近散架?
如果这一步让分子变得更稳定、更符合物理规律,AI 就得到一个「奖励」,并记住这是个好操作。
如果这一步让分子变得更不稳定,AI 就受到一个「惩罚」,并学会避免这条路。
经过这种持续的物理反馈与奖惩循环,AI 在创作的每一步都必须思考物理后果,超越了单纯模仿训练数据中分子形态的局限。
效果如何?
经过「家教」调教的 AI,生成的分子质量上了一个台阶。
在 QM9 标准数据集上,它生成的分子中,物理稳定性的比例从 82% 提升到了 93% 以上。
这个「家教」框架具备通用性,能指导 EDM、GeoLDM、UniGEM 等不同的扩散模型。它是一个普适的、可即插即用的「增强模块」。
这项工作跳出了比拼模型参数的范式,代表了一种根本性的思路转变。它将物理学知识注入到 AI 的生成过程中。
📜Title: Guiding Diffusion Models with Reinforcement Learning for Stable Molecule Generation
📜Paper: https://arxiv.org/abs/2508.16521
💻Code: https://github.com/ZhijianZhou/RLPF/tree/verl_diffusion
3. DrugReasoner:能解释自己的 AI 预测
药物发现领域充斥着各类 AI 预测模型,它们可以预测分子的溶解度、毒性或结合亲和力。但很少有人敢将数亿美元的临床试验,完全押注于一个 AI 模型给出的数字。
因为这些模型大多是黑箱。
当一个模型给出 0.87 的成药性得分,却无法解释原因时,它就像一个只报数字的算命先生。决策者无法判断预测是基于严谨计算还是随机猜测,这种无法解释的结果,应用价值有限。
DrugReasoner 的设计正是为了解决这个痛点。
AI 学会了「举一反三」
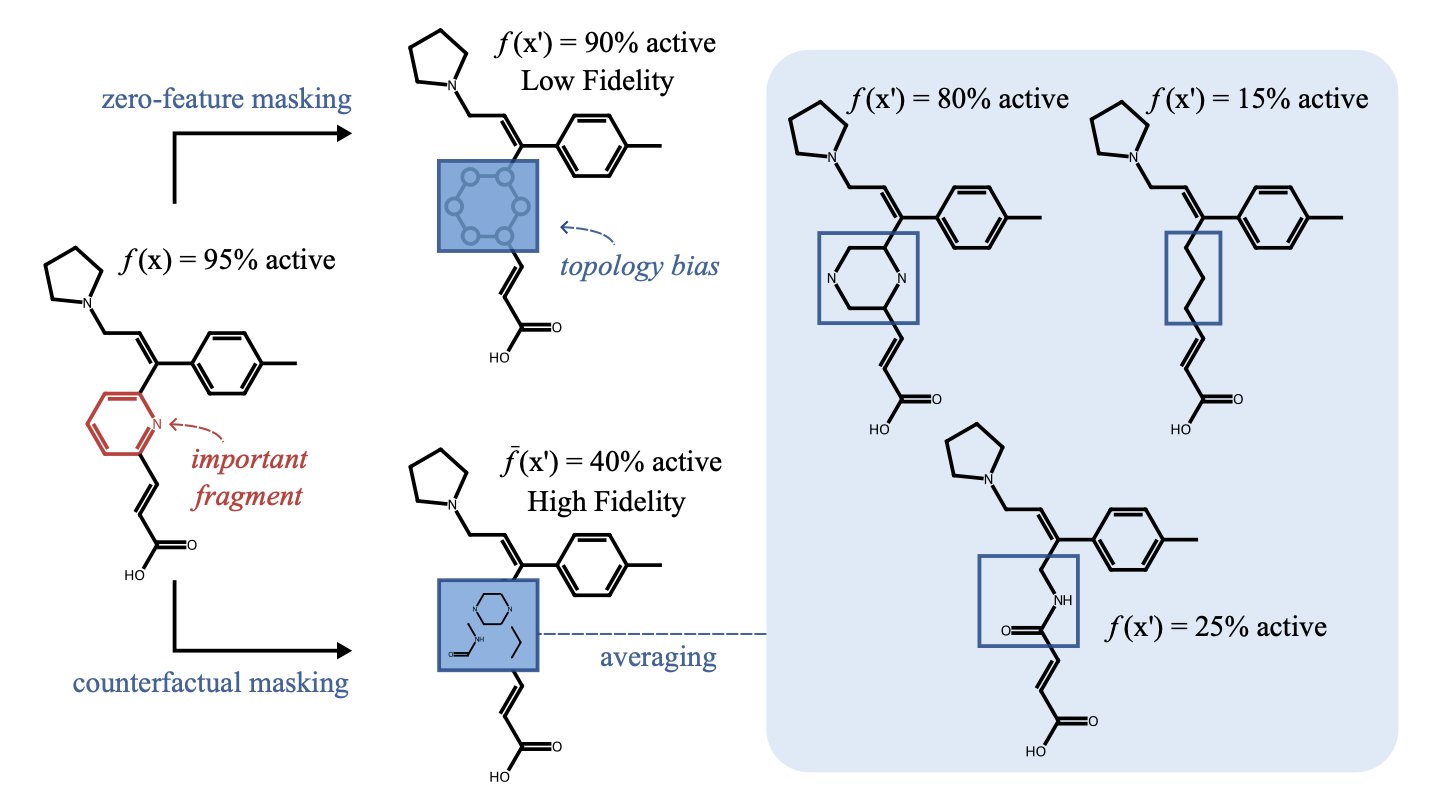
研究者训练 AI 掌握了化学家的一项核心技能:比较。
当输入一个新设计的候选药物时,DrugReasoner 会检索知识库,找出结构最相似的已知药物。随后,它将这些相似药物分为两组:一组是已成功上市的,另一组是在临床试验中失败的。
最后,它生成一份推理报告,例如:
「此分子有 73% 的概率获批。其 A 部分结构与某成功抗癌药相似,该结构通常成药性良好。但其 B 部分结构与某因肝毒性失败的分子类似,这会降低其获批概率。」
这种解释方式提供了可用于决策的清晰信息。
结果怎么样?
在包含大量真实药物分子的数据集上测试,DrugReasoner 的预测准确率(AUC 为 0.73)与传统的机器学习方法(如 XGBoost)相当,并优于另一新近发布的模型 ChemAP。
这表明,模型在提供可解释性的同时,并未牺牲预测准确性。
预测新药能否获批,受科学、商业、运气等多种变量影响。在此背景下,AUC 0.73 是一个可靠的成绩,但并非完美无缺。
研究者承认,为防止模型通过记忆 SMILES 字符串「作弊」,训练时未提供分子的 SMILES 表示。这或许限制了模型对精细化学结构的理解,但也证明了该推理方法在信息受限的情况下依然有效。
📜Title: DrugReasoner: Interpretable Drug Approval Prediction with a Reasoning-augmented Language Model
📜Paper: https://arxiv.org/abs/2508.18579
💻Code: https://github.com/mohammad-gh009/DrugReasoner
4. Polaris 挑战赛:用碎片知识提升对接精度
一线从事结构药物设计的研究者对分子对接软件的感情很复杂。它的速度快,能在一夜之间筛选百万级分子库。但它的打分函数又常不可靠。软件会生成大量看似理想的结合姿态并给出高分,但实验验证的结果却往往不符。
对接软件就像一个速度飞快但鲁莽的侦察兵,能迅速勘察整个战场,却也常把稻草人误报为敌人。研究者需要的是一副「敌我识别」眼镜。Polaris 挑战赛的一篇论文展示了这样一副眼镜。
别再让 AI 瞎猜了,给它一本「小抄」
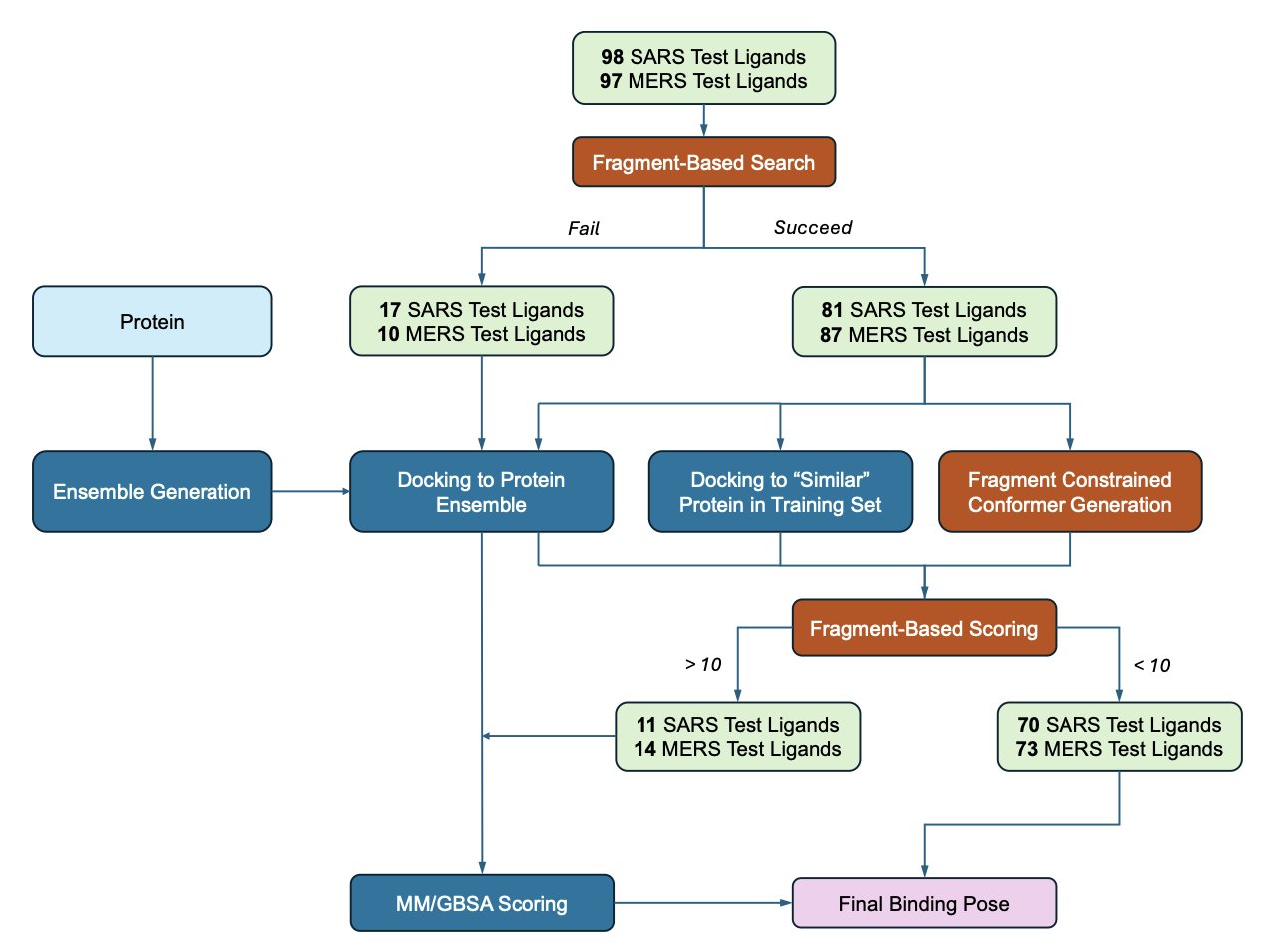
研究者的思路并非重造对接引擎,而是改进打分环节。他们认为 Vina 等工具在生成多样化姿态方面已足够好,短板在于打分。
他们使用的「小抄」是数据库中成千上万个小分子片段的晶体结构。
这些片段如同「路标」,通过实验数据标示出蛋白质口袋中哪些区域疏水、哪些位置适合形成氢键。
新流程首先使用 VinaGPU 生成大量可能的结合姿态。随后,AI 利用「小抄」进行批改:检查分子中的基团是否与已知片段的「路标」位置匹配。若匹配则加分,反之则减分。
流程还以更主动的方式利用这份「小抄」:约束对接。如同考前提示,它能引导对接程序从初始阶段就朝正确的方向搜索。
如果没有小抄怎么办?
当缺乏可参考的片段数据时,流程会启用后备方案:MM/GBSA重打分。
这种基于物理学的传统计算方法虽然比 Vina 的打分函数慢,但通常更准确。
整个流程构成一个层层递进的过滤器:用最快的方法海选,用数据驱动的知识精选,最后用物理学方法复核。
这东西真的靠谱吗?
研究者将该流程应用于新冠病毒(SARS-CoV-2)及其近亲中东呼吸综合征冠状病毒(MERS-CoV)的蛋白酶。
结果显示,对超过一半的分子,该流程成功预测出与晶体结构误差在 2 埃以内的结合姿态。这在真实世界靶点上是一个可靠的成绩。
他们还证明,从 SARS-CoV-2 片段数据中学到的知识,可以成功迁移并用于 MERS-CoV 的预测。
这项工作提供了一个可部署的工程解决方案,旨在解决日常研究中的实际问题。计算药物发现的未来,在于巧妙地组合数据、物理学原理与化学直觉,而非依赖单一工具。
📜Title: Polaris Challenge: Data-driven Priors to Improve Docking for Pose Prediction
📜Paper: https://doi.org/10.26434/chemrxiv-2025-fv3c3
5. DRUGFLOW:AI 学会了画「藏宝图」
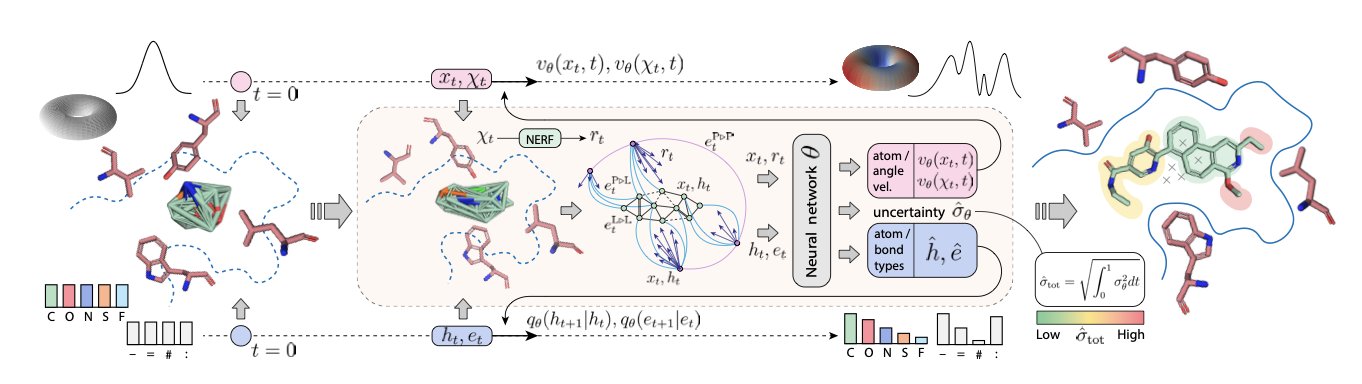
- 它学习整个「好分子」的分布,如同绘制一张完整的藏宝图,以此取代只标记单个宝藏点的旧方法。
- 它内置了不确定性检测、蛋白质柔性等化学家需要的功能,让 AI 从艺术家向工程师转变。
与生成式 AI 打过交道的人都清楚,AI 是顶级的艺术家,却是二流的化学家。给它一个蛋白质口袋,它能「画」出结构新颖、看似完美的分子。但这些分子大多只是在统计上「貌似」已知药物,AI 并未真正理解一个分子「好」在何处。
就像一个只会临摹梵高画作的画匠,他能画出星空,却不懂其深意。
DRUGFLOW 的目标,就是教 AI 去学天文学。
从「画快照」到「绘地图」
这项工作的核心思路发生了转变。一个好药物本身就坐落在广阔「化学空间」里,是一片能量低、性质好的区域。因此,AI 的任务是学会绘制出这片区域的完整「地图」,取代以往生成单张「快照」的做法。
DRUGFLOW 通过结合两种数学工具——连续流匹配(Continuous Flow Matching),可想象为用平滑曲线描绘地形;以及离散马尔可夫桥(Discrete Markov Bridges),可想象为在关键地标间建立逐步路径——来学习蛋白质口袋里所有「好分子」的完整分布。
学会绘制地图后,AI 便从模仿的画匠,转变为能指出山峰与盆地的地理学家。
AI 终于长了点「脑子」
拥有全局观后,AI 便能实现一些过去难以企及的功能。
首先是不确定性。DRUGFLOW 内置了「不靠谱探测器」。当它生成的分子落在地图上未曾探索的区域,它会直接提示:「我不确定这个分子是否可靠。」这种知道自己「不知道」的能力,能避免合成 AI 幻想出的分子。
其次是尺寸自适应。生成分子时,它会动态调整大小,主动删除可能与蛋白质口袋产生空间位阻的原子。就像装修工把沙发塞进门前,会先拆掉沙发腿。
最后是蛋白质柔性。升级版 FLEXFLOW 在生成配体分子的同时,能同步调整蛋白质口袋的氨基酸侧链朝向。真实世界里,蛋白质口袋并非坚硬的模具,而像一只柔软的手套。FLEXFLOW 如同一个真正的设计师,在设计手的同时,能让手套自适应手的形状。
📜Title: Multi-Domain Distribution Learning for De Novo Drug Design
📜Paper: https://arxiv.org/abs/2508.17815