
目录
- CAML 通过强迫 AI 不仅要看一个蛋白质的「简历」(序列和结构),更要看它的「住址」和「邻居」(基因组语境),从而极大地提升了功能预测的准确性。
- Q-MOL 不再将蛋白质视为僵硬的「锁」,而是将其建模为一张柔性的「能量地形图」,从而能发现那些隐藏在平坦表面的变构「山谷」,并在真实世界的筛选中,取得了惊人的 36% 命中率。
- 这篇综述点明了药物联用预测的未来:AI 必须学会看懂蛋白三维结构这个「物理战场」,而不是只盯着信号通路那张二维「作战地图」。
1. AI 蛋白功能预测:学会看「邻居」的重要性
在功能基因组学中数据分析师每天都在扮演着一种分子级别的「人力资源经理」的角色。一个新测序出的宏基因组,会扔给你成千上万个从未见过的、功能未知的「应聘者」(蛋白质)。我们手头,通常只有它们的「个人简历」——也就是它们的氨基酸序列,以及用 AlphaFold 预测出的、质量参差不齐的「证件照」(三维结构)。然后,我们需要仅凭这些,去猜测这个蛋白质,到底是个激酶,还是个转运蛋白,还是别的什么东西。
过去,我们所有的 AI 模型,基本上也都在干同样的事。它们是无比强大的「简历筛选器」。它们能从序列和结构里,读出很多深刻的信息。但它们一直以来,都忽略了一个做生物学的人,凭直觉就知道无比重要的信息:这个应聘者,他住在哪?他的邻居,都是干什么的?
CAML 想给这位「HR 经理」,配上一套最先进的「背景调查」工具。
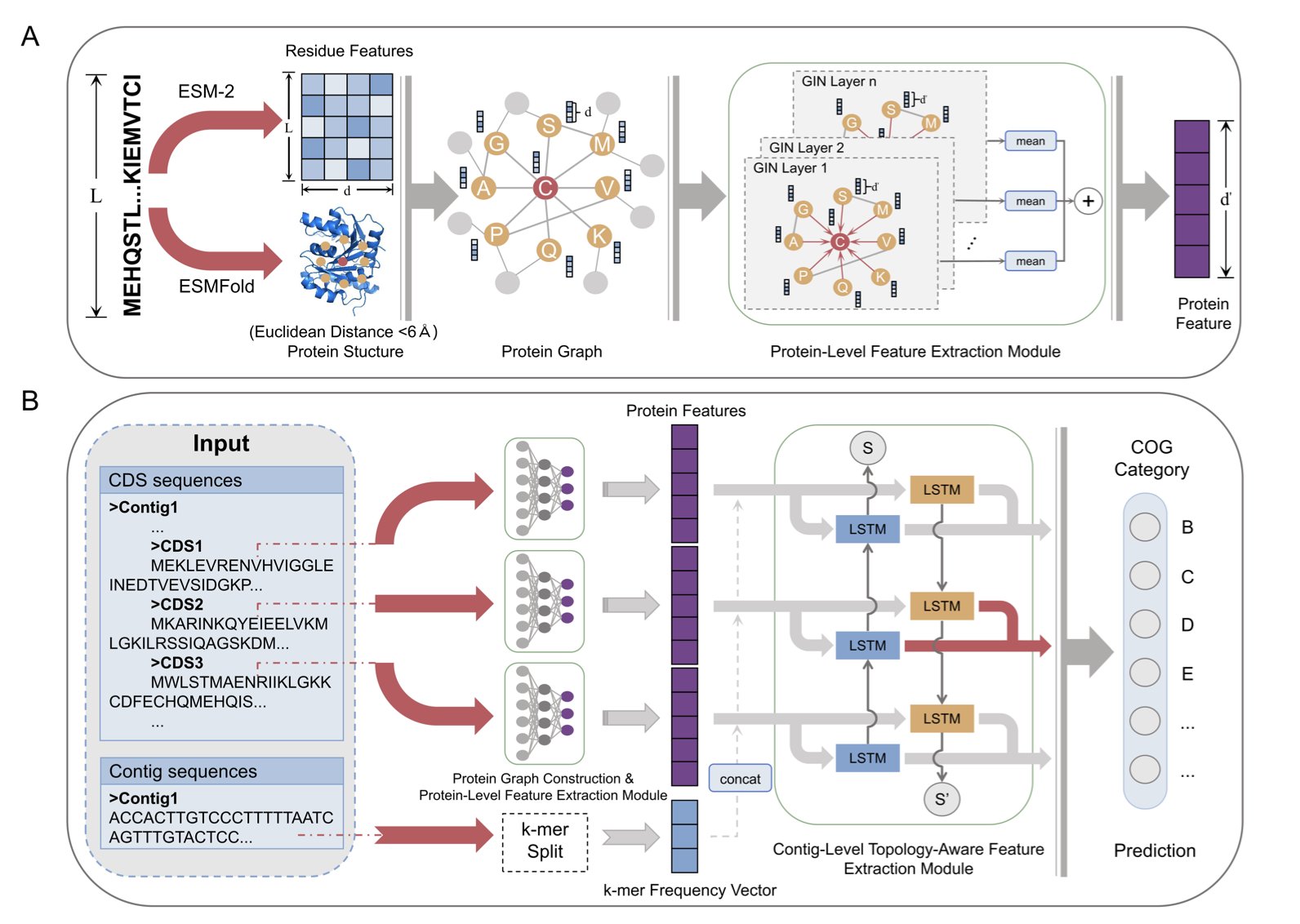
CAML 的架构,像是一个全面的招聘流程:
1. 第一步:看简历。
用的是我们这个时代的「梦之队」。它用 ESM-2 这个「语言学大师」,去「阅读」蛋白质的氨基酸序列;同时,它用一个图同构网络(GIN),去「审视」蛋白质的接触图谱,也就是它的三维结构。到这里,它已经把一个蛋白质的「个人素质」,给摸了个底朝天。
第二步:进行背景调查。
CAML 的点睛之笔。它会去看这个基因,在它的染色体片段(contig)上,都和哪些基因做「邻居」。这在微生物的世界里,是一个无比强大的线索。因为功能上相关的基因,常常会被组织在一起,形成我们称之为「操纵子」的「功能社区」。这就像是你发现一个应聘者,他住的那条街上,全是顶级的软件工程师。那你就有很强的理由怀疑,他就算不是个程序员,也至少是个产品经理。CAML 用一个双向长短期记忆网络(BiLSTM),去「阅读」这种基因邻里关系,从而为这个蛋白质,建立起一个「社会关系」画像。第三步:综合面试,做出最终判断。
最后,CAML 会把这个蛋白质的「个人简历」和它的「背景调查报告」,放在一起,进行一次最终的、综合的评估,从而给出它关于这个蛋白质功能的、最可靠的判断。
那么,同时看了「简历」和「背景调查」的新 HR 经理,表现如何呢?
它把所有只看简历的「老前辈」们,都给比了下去。而且不是只好了一点点。在准确率和 F1 分数上,它的提升,动辄就是 50%、60%。而「消融实验」证明了,这个引擎的燃料,主要就来自于过去一直忽略的、宝贵的「基因组语境」信息。
📜Title: Protein Function Prediction via Contig-Aware Multi-Level Feature Integration
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.07.669053v1
2. Q-MOL:给蛋白质做「地形图」来寻找药物
计算辅助药物设计一直活在一个经典的、但越来越显得力不从心的比喻里:「锁和钥匙」。
我们把蛋白质,当成一把形状固定的、僵硬的「锁」;把药物分子,当成一把同样坚硬的「钥匙」。然后,我们所有的计算,都是在尝试,看这把钥匙,能不能插进这把锁里,以及插得有多「严丝合缝」。
但我们都清楚,这不是事情的全貌。
蛋白质,它不是一块花岗岩。它更像是一个由无数个微小的、互相连接的弹簧和铰链组成的、一直在微微晃动和呼吸的机器。有时候,一个药物分子,需要先轻轻地「敲一敲门」,蛋白质才会为它打开一个本来不存在的「隐藏口袋」。我们称之为「诱导契合」。而那些传统的、把蛋白质当成「刚体」的对接程序,在面对这种动态的复杂性时,基本上就束手无策了。更别提那些本身就像一团煮熟的意面的「本质无序蛋白」了。
Q-MOL 试图用一个更接近物理学现实的思路来解决这个问题。
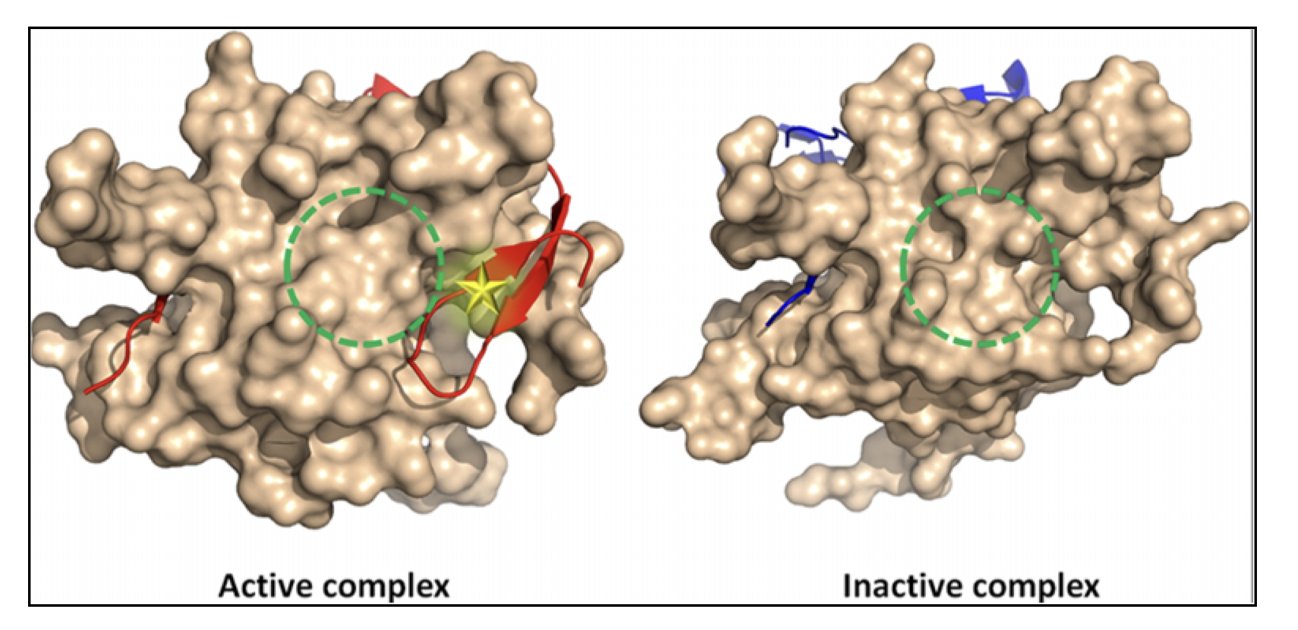
它采取了一种「配体为中心」的视角。它不再问:「这把钥匙,能不能插进这把锁?」它问的是:「对于这把钥匙来说,在这把锁的整个表面上,哪个地方,是它待着最舒服、能量最低的‘风水宝地’?」
为了回答这个问题,Q-MOL 把整个蛋白质受体,不再看成一个固定的三维结构,而是看成一个多维的「势能景观」。你可以把这,想象成一张无比详尽的、包含了山峰、峡谷、平原和盆地的「地形图」。这张地形图,就隐式地包含了蛋白质所有可能的、低能量的柔性构象。
现在,对接过程,就变成了一个非常直观的物理过程。Q-MOL 把那个配体分子,像一个玻璃弹珠一样,放在这张「地形图」上。然后,它就让这个弹珠,在重力的作用下,自由地滚动,直到它最终停在某个能量最低的「山谷」里。
这个「山谷」,可能就是我们早已知道的、那个经典的活性位点。但它,也可能是一个我们从未见过的、在蛋白质表面一个看似平坦的区域里、一个微小的、隐藏的「洼地」——也就是我们梦寐以求的「变构口袋」。
那么这在现实世界里管用吗?
他们把这套系统,用在了西尼罗河病毒的一个蛋白酶上。这是一个真实的、困难的药物靶点。他们让 Q-MOL,从一个虚拟化合物库里,挑出了 50 个它认为最有希望的分子。然后,他们真的,回到实验室,把这 50 个分子,都给测试了一遍。结果,其中有 18 个,显示出了真实的抑制活性。
36% 的命中率。
他们还把这个平台,用在了像 cMyc 和β-catenin 这样的、出了名的、「不可成药」的靶点上,并成功地预测出了潜在的变构结合位点。
📜Title: Q-MOL: High Fidelity Platform for In Silico Drug Discovery and Design
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.06.668254v1
3. AI 预测药物组合:从通路图到 3D 战场
在药物研发中,尤其是肿瘤领域,我们都梦想着能打出「1+1>2」的协同组合拳。但现实往往是,我们花了大价钱,最后打出的是「1+1<1」的拮抗组合,甚至还附赠一堆毒副作用。
为什么?
因为我们过去预测药物组合效应的方式,太像是在看一张简化的地铁线路图。
我们看着信号通路图,说:「药 A 堵住了 A 站,药 B 堵住了 B 站,那这条线路就瘫痪了!」但我们完全忽略了,细胞内不是只有几条孤立的地铁线。它是一个拥挤、混乱、充满了各种「换乘」和「地面交通」的立体城市。
这篇综 - 述,就是要把我们的目光,从那张扁平的「地铁图」,拉回到这个三维的、动态的「城市」本身。它大声疾呼:别只看通路,看结构!
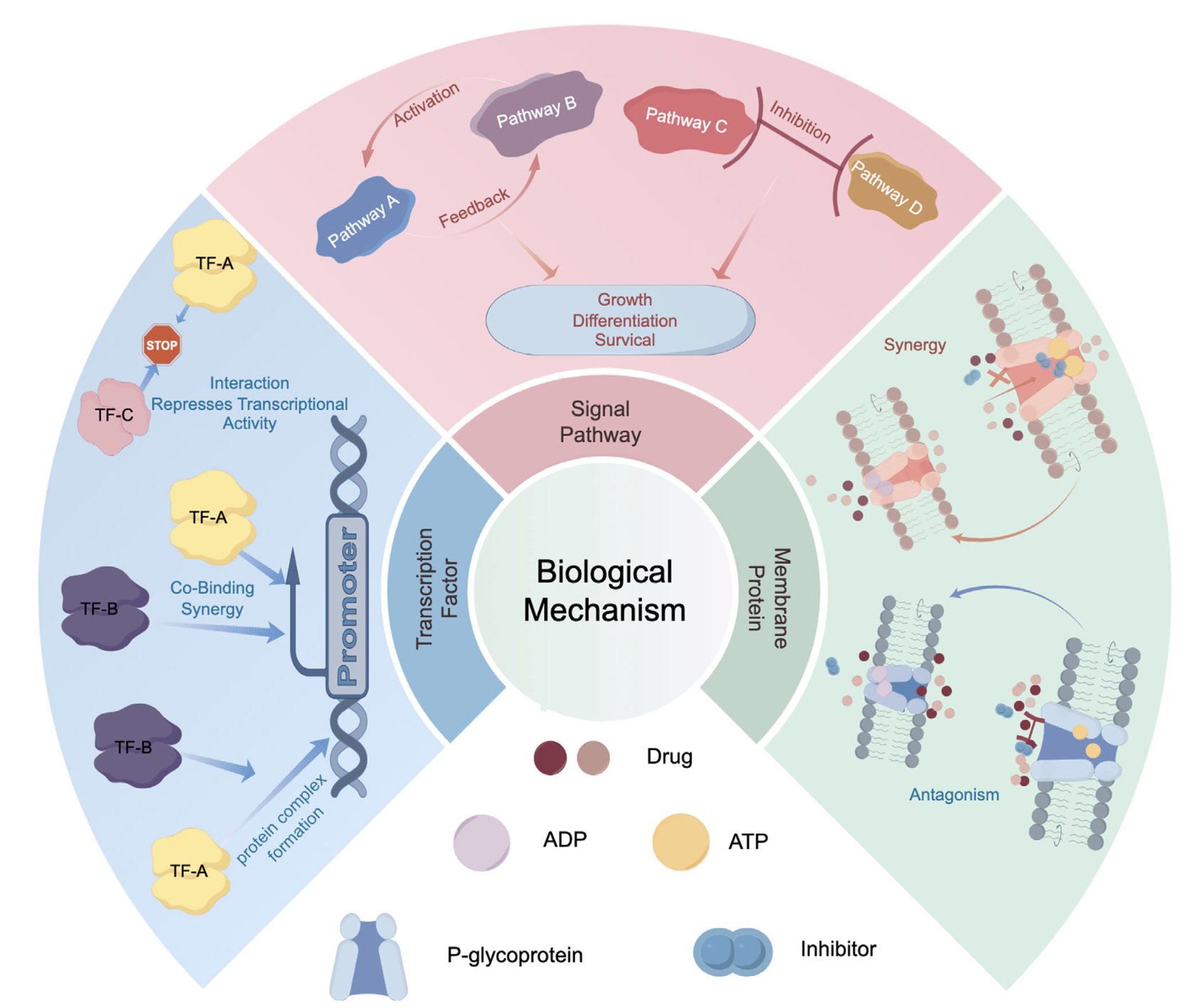
它的核心论点非常符合物理直觉。药物协同或拮抗的根本原因,不是抽象的箭头,而是具体的分子「握手」。
* 协同作用可能是什么样的?也许药物 A 结合到蛋白 X 上,像一把钥匙一样,把蛋白 X 拧成了一个新的形状。而这个新形状,恰好就暴露出了一个完美的、过去不存在的结合口袋,让药物 B 能以极高的亲和力结合上来。
* 拮抗作用呢?可能药物 A 让蛋白 X 变得「害羞」,缩成一团,把原本药物 B 的结合位点给藏了起来。
在 AlphaFold 和各种结构预测 AI 出现之前,要系统性地思考这种分子级别的「蝴蝶效应」,简直是天方夜谭。而现在,AI 给了我们一个机会,去当这场复杂分子芭蕾舞的「编舞导演」。这篇综述系统地梳理了,如何利用 AI,去分析药物如何改变蛋白的构象系综,如何影响蛋白 - 蛋白相互作用的界面,如何竞争同一个转运蛋白的「通道」。
好了,梦想很美好,该泼冷水了。
这篇综述也指出了那个「房间里的大象」——数据。
我们哪儿来那么多高质量的训练数据?我们连单个药物和蛋白相互作用的可靠数据都还不够多,更别提药物组合了。要去系统性地测量,上百种药物组合,如何影响细胞内成千上万种蛋白的动态和功能,这在实验上是一个浩瀚如星辰大海的、足以让任何一个实验科学家望而生畏的工程。
📜Title: Protein Spatial Structure Meets Artificial Intelligence: Revolutionizing Drug Synergy–Antagonism in Precision Medicine
📜Paper: https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202507764