
目录
- GP-MOBO 利用高斯过程和 Tanimoto 核,在不牺牲分子指纹完整性的前提下,高效地进行多目标贝叶斯优化,为药物化学家提供了更靠谱的分子设计导航。
- LABind 创新地在预测蛋白质结合口袋时,将配体信息也纳入考量,让预测不再是「盲猜」,而是能根据不同配体的「长相」来找到最适合它的「座位」。
- B3clf 通过整合重采样技术来解决血脑屏障预测中常见的数据不平衡问题,最终打包成一个开箱即用的工具,为中枢神经系统药物研发提供了务实的帮助。
1. GP-MOBO:更懂化学家的多目标分子优化
在做药物发现时,分子优化是个永恒的主题。我们总是在玩一个「权衡」的游戏:活性要高,溶解度要好,毒性要低,代谢要稳定……这些目标常常是相互矛盾的。你把分子做得更脂溶一点,活性可能上去了,但溶解度又掉下来了。
贝叶斯优化(BO)是解决这类多目标优化问题的利器。你可以把它想象成一个勘探队员。它不会把整个山头都挖一遍,而是先在几个点打钻取样,然后根据这些样本,建立一个关于地下矿藏分布的「概率地图」(高斯过程模型)。接着,它会根据这个地图,去预测下一个最有可能挖到宝藏的点。
但传统的贝叶斯优化,在用到化学世界里时,经常会遇到一个问题:它不太「懂」化学。
问题的根源:高斯过程的「化学盲」
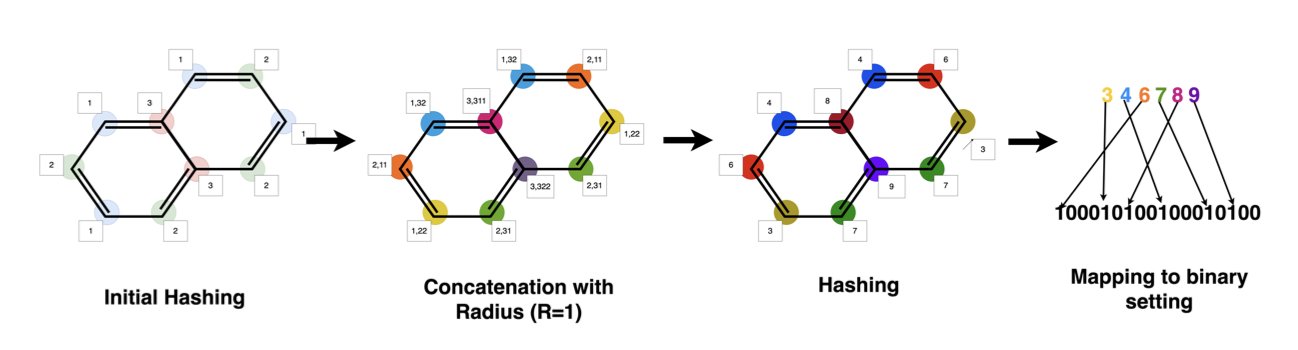
我们描述一个分子,最常用的方法是「分子指纹」(fingerprints)。这就像一个长长的 0 和 1 组成的条形码,记录了这个分子有哪些化学基团、哪些结构特征。这种表示方法有个特点:维度很高(几千维),而且极度稀疏(大部分都是 0)。
传统的高斯过程模型,在这种高维稀疏的数据上,表现往往不佳。它很难理解两个分子的「化学相似性」。在它眼里,两个指纹上只有几个比特位差异的分子,可能和差异巨大的分子没什么区别。这就导致它的「概率地图」画得不准,给出的下一步建议也常常不靠谱。
GP-MOBO 的解法:教高斯过程学化学
这篇论文的 GP-MOBO 算法,就是给高斯过程换了一个更懂化学的「大脑」——Tanimoto 核。
Tanimoto 系数是化学信息学里衡量分子相似性的金标准。把 Tanimoto 系数的思想做成一个「核函数」,再把它整合进高斯过程里,就相当于教会了那个勘探队员用化学家的眼光去看待分子。
现在,当模型看到两个分子的 Tanimoto 相似性很高时,它就会推断,这两个分子的性质也应该很接近。这样一来,它建立的「概率地图」就远比之前要精准。它能更好地理解 SAR(构效关系),从而给出更靠谱的优化方向。
实际效果如何?
作者们在 DockSTRING 这个真实世界的分子对接数据集上,把 GP-MOBO 和传统的 GP-BO 方法进行了对比。结果显示,在仅仅 20 轮的优化迭代中,GP-MOBO 找到的「帕累托前沿」上的分子,质量和多样性都显著更优。
「帕累托前沿」是什么?可以把它想象成所有「没有被完全碾压」的候选分子的集合。在这个集合里,任何一个分子,你都找不到另一个分子能在所有优化目标上都比它好。对于药物化学家来说,我们需要的不是一个「单项冠军」,而是这个前沿上的一系列各具特色的「全能选手」,以便我们根据其他未能量化的因素(比如合成难度)来做最终决策。
GP-MOBO 能给我们提供一个更丰富、更高质量的「候选名单」,这就是它最大的价值。它没有用什么复杂的深度学习模型,而是回归到经典的统计方法上,通过一个巧妙的、更符合化学原理的核函数,解决了分子优化中的一个关键痛点。
📜Paper: https://arxiv.org/abs/2508.14072
2. LABind:一个会看「配体脸色」的结合口袋预测 AI
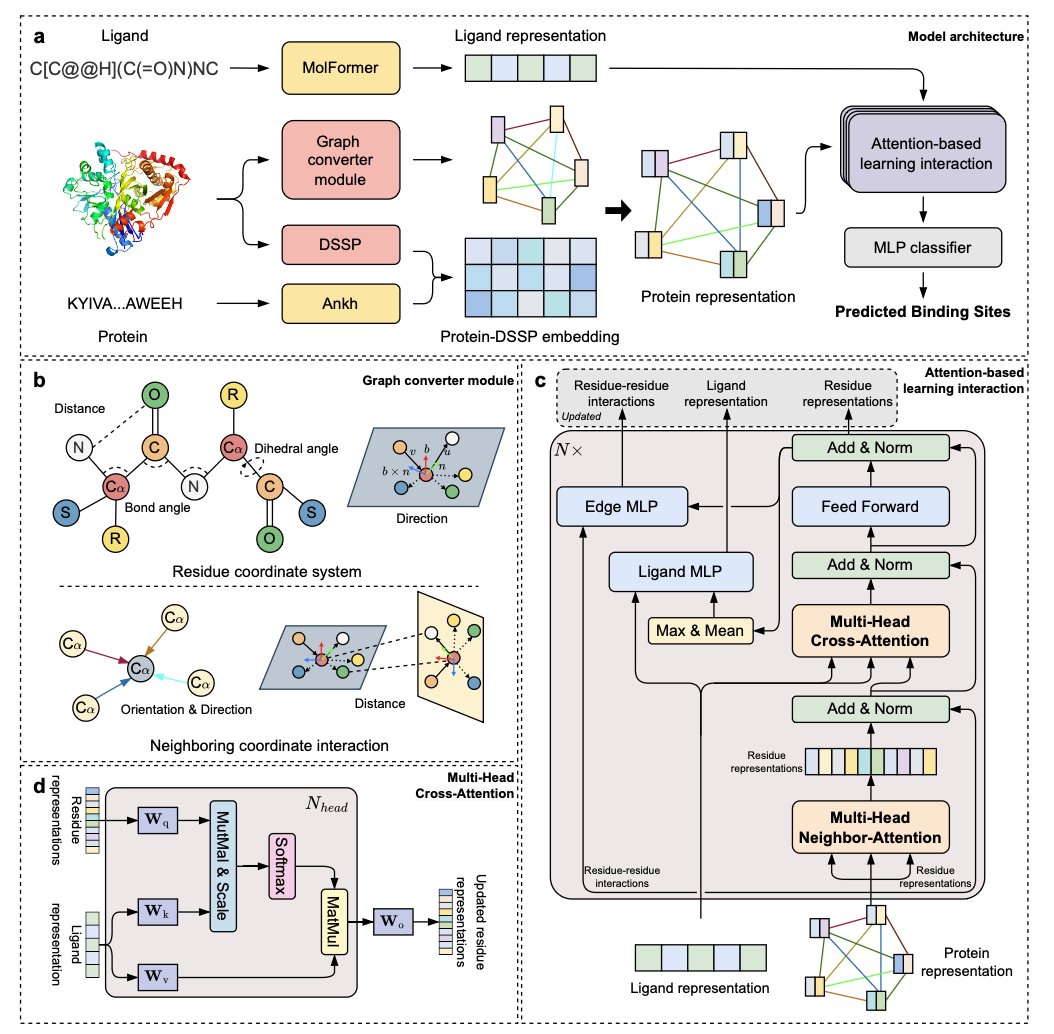
在做药物发现时找到小分子配体在靶点蛋白上的结合口袋,是所有工作的第一步。过去,我们有很多方法来预测口袋,大部分方法是「配体无知」的(ligand-agnostic)。它们只分析蛋白质的表面,寻找那些形状和物理化学性质适合小分子结合的「坑」或「洞」。
这种方法在很多时候管用,但它有一个根本性的问题:它没有考虑到,不同的配体,可能喜欢待在同一个蛋白质的不同位置。一个又大又油腻的分子,和一个又小又带电的分子,它们心仪的「座位」可能完全不同。只看蛋白质而不看配体,就像一个餐厅服务员,不问客人喜欢什么口味,就随便给客人安排座位一样。
LABind 是怎么做到「看人下菜碟」的?
LABind 的核心,是一个叫「交叉注意力机制」(cross-attention mechanism)的东西。你可以把它想象成「服务员」的一双眼睛。
然后,最关键的一步来了。交叉注意力机制,会让模型去反复地比较这两个「编码」。它会学习去发现它们之间的「匹配模式」。比如,模型会慢慢学会:「哦,当配体上有个带负电的羧基时,蛋白质上那个带正电的赖氨酸附近,就是一个很好的结合位点。」
通过学习成千上万个已知的蛋白 - 配体复合物结构,LABind 就逐渐掌握了这种根据配体来动态调整口袋预测的「直觉」。
泛化能力才是硬道理
一个模型在训练集上表现好不稀奇,真正的考验是,当给它一个它从未见过的蛋白质,或者一个化学骨架全新的配体时,它还能不能给出靠谱的预测。这就是我们说的「泛化能力」。
LABind 在这方面的表现非常出色。在多个基准数据集的测试中,它都优于现有的其他方法,尤其是在预测全新配体的结合位落时。
更实用的一点是,我们现在有很多蛋白还没有实验解析出的三维结构。LABind 可以直接使用像 ESMFold 这类 AI 工具预测出的蛋白结构,依然能保持不错的预测精度。这就大大拓展了它的应用范围,让我们能去探索更多未知的靶点。
文章中还展示了一个非常及时的案例:预测新冠病毒 NSP3 蛋白与不同抑制剂的结合口袋。即使是对于那些全新的、模型在训练中没见过的抑制剂,LABind 也给出了准确的预测。
LABind 把口袋预测,从一个「静态的」找坑游戏,变成了一个「动态的」匹配问题。它让我们能更精准地回答「这个特定的分子,最有可能结合在这个蛋白的什么位置?」这个问题。这对于虚拟筛选、分子对接以及理解药物作用机制,都是一个非常强大的新工具。
📜Paper: https://www.nature.com/articles/s41467-025-62899-0
💻Code: https://github.com/ljquanlab/LABind
3. B3clf:一个更靠谱的血脑屏障预测开源工具
做 CNS 药物的绕不开血脑屏障(BBB)。合成一个新分子,除了要看它对靶点的活性,最头疼的就是问:「它到底能不能进脑?」这个问题要是没谱,后面全是白费功夫。
所以,多年来我们一直依赖各种计算模型来做预测。但用过的人都知道,这些模型的准确率时好时坏,有时候感觉还不如老药化专家的直觉靠谱。
为什么会这样?算法不够好吗?不全是。一个更根本的问题,出在训练模型用的数据上。
问题的根源:倾斜的数据天平
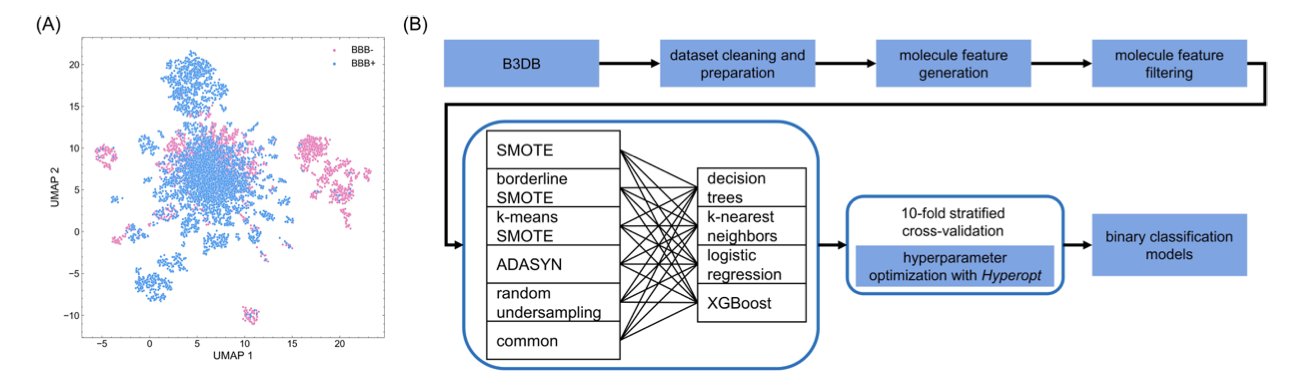
你想想看,在公开的数据库里,能顺利通过血脑屏障的分子是少数,大部分都惨遭拦截。这就造成了典型的「类别不平衡」问题。
这意味着什么?一个机器学习模型如果想偷懒,它可以学会一个最简单的策略:对所有分子都预测「不能通过」。这样做,它的整体准确率可能还挺高,比如能达到 80% 甚至 90%,因为它猜对了大部分情况。但这种模型对研发人员来说,一点用都没有。我们真正想找的,是那少数能进去的「天选之子」,而模型恰恰放弃了学习如何识别它们。
这篇论文直面了这个最 t 棘手的数据问题。
B3clf 是怎么做的?
他们的思路很直接:既然天平是斜的,那就想办法把它扶正。他们用的方法叫「重采样」(Resampling)。
你可以这么理解:对于数据量少的那一类(能通过 BBB 的分子),我们不能只是简单地把它们复制几遍,那样模型还是学不到新东西。像 ADASYN 或 SMOTE 这类更智能的过采样技术,它会在已有的「好学生」周围,创造出一些新的、看起来也很像「好学生」的「虚拟样本」。这样一来,模型就有了更丰富、更均衡的学习材料,能更好地理解一个分子到底需要具备哪些化学特征(比如合适的脂溶性、分子量、氢键数量等)才能拿到进入大脑的「通行证」。
不搞个人崇拜,让实力说话
作者们没有上来就认定哪个模型最好。他们做了一件事:搞了一场「模型大比武」。他们挑选了 24 种不同的机器学习模型,从简单的决策树到复杂的 XGBoost,让它们分别与不同的重采样技术配对组合,然后在同一个数据集上跑分。
结果显示,XGBoost(一种梯度提升树模型)在这次比赛中表现最突出,尤其是在和过采样技术结合后,无论是在准确率、灵敏性还是其他关键指标上,都名列前茅。XGBoost 本身就是机器学习领域里出了名的「优等生」,稳定又强大,这个结果并不意外,但通过这样系统性的比较,让结论更有说服力。
是骡子是马,拉出来遛遛
光说不练假把式。他们还从文献里整理了一个包含 175 个化合物的外部测试集,用来和其他已发表的 BBB 预测模型进行正面 PK。结果表明,B3clf 训练出的模型性能不输于,甚至优于现有的一些方法。
他们把训练好的模型打包成了一个开源的 Python 库和一个网页工具。这意味着,我们不需要自己去折腾代码和数据。开项目会的时候,我们可以直接把新设计的几个分子的 SMILES 串贴进去,几秒钟就能得到一个能否通过 BBB 的预测概率。这个概率值可以帮助我们决定,下一批该优先合成哪个分子。
📜Paper: https://doi.org/10.26434/chemrxiv-2025-xschc
💻Code: https://github.com/theochem/B3clf