
目录
- Schrödinger 新发布的 Glide WS,通过显式处理结合位点中的水分子,显著提升了对接准确性和虚拟筛选效率,这可能是对传统刚性受体对接方法的一次重要升级。
- 研究者们用神经网络当向导,让数据自己说话,迭代式地挖出分子从 A 到 B 最可能的“高速公路”,解决了反应路径搜索的一大痛点。
- MEVO 巧妙地将分子生成与进化策略结合,用海量‘无主’配体数据训练模型,为结构化药物设计开辟了一条新路。
- 通过将化学知识“原子化”为官能团,ChemDFM-R 让大模型不再是鹦鹉学舌,而是真正开始理解化学反应的底层逻辑了。
- LLM 能大幅简化合成健康数据的创建流程,但它是个强大的副驾驶,而不是能取代人类专家智慧的自动巡航系统。
1. Glide WS 发布:水分子,分子对接的新战场
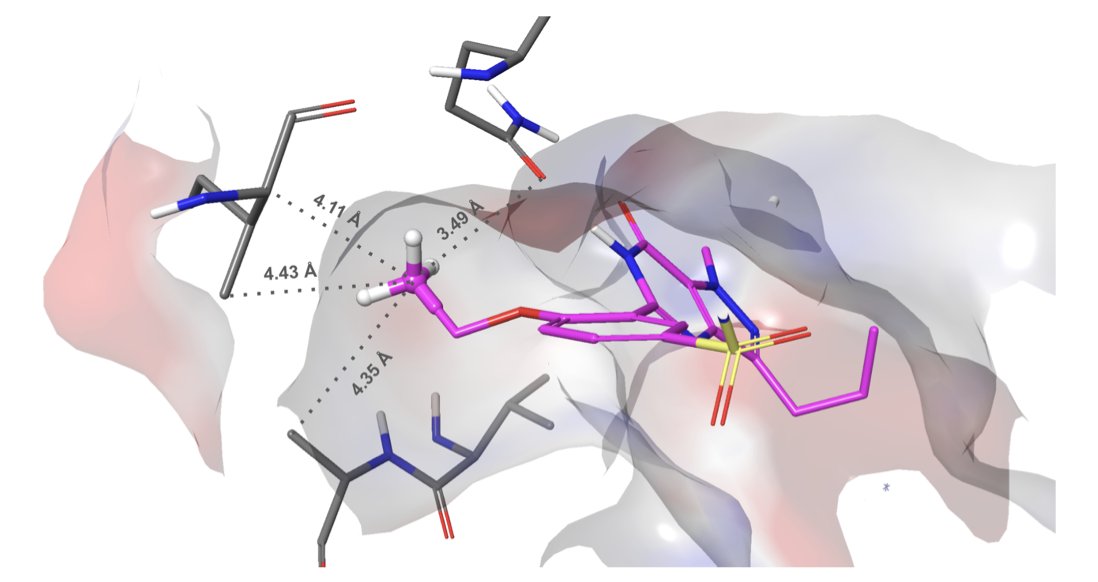
做分子对接这么多年,我们心里都清楚一个“房间里的大象”:水。
我们总是假装那个漂亮的蛋白口袋是真空的,然后把小分子塞进去打分。但现实呢?口袋里全是水分子,它们上蹿下跳,形成复杂的氢键网络,既是配体结合的“路障”,也是可以利用的“跳板”。
一直以来,怎么处理这些水,始终是分子对接领域一个老大难的问题。大多数程序要么干脆忽略,要么用一些粗糙的隐式溶剂模型糊弄一下。
现在,Schrödinger 带着 Glide WS 来了,声称他们终于正面解决了这个问题。这可不是小打小闹的升级。
他们没用什么玄学,而是祭出了自家另一个工具 WaterMap。简单来说,就是在对接开始前,先用分子动力学模拟(MD)算一遍,看看蛋白口袋里的哪些水分子待得“不舒服”(能量高),哪些待得很安逸(能量低)。然后,Glide WS 的新评分函数就学会了“看水下菜”:如果你的配体能恰好赶走那些“不舒服”的水分子,占据它的位置,那你就能得到一个大大的奖励分。反之,你要是想挤走一个开开心心的水,那就得付出能量代价,扣分。
这套逻辑听起来就比以前的“去溶剂化惩罚”要精妙得多。效果怎么样?
在一个包含近 1500 个复合物的广泛测试集上,Glide WS 的自对接(self-docking)准确率达到了 92%,而之前的基准 Glide SP 则为 85%。在对接领域,7% 的绝对提升,特别是在 85% 这样高的基准上,确实算得上是巨大的进步。这意味着我们得到的结合模式(binding pose)更有可能是正确的,这对所有后续基于结构的药物设计工作来说,都是基础性的利好。
当然,算得准不如筛得准。虚拟筛选的本质就是从成千上万的分子大海里捞针。Glide WS 在这方面的表现更让人兴奋。
报告里说,它能显著降低那些“伪高分”的干扰分子(decoys)的数量。也就是假阳性率大大降低了。这意味着什么?这意味着我们不用再浪费时间和金钱去合成、测试一大堆其实根本没活性的化合物。以前可能要测试 1000 个分子才能找到一个苗头,现在也许只要测 200 个。这对于项目进度和预算来说,简直是福音。
更有意思的是,他们把一些药物化学家的“祖传经验”也塞进了评分函数。比如那个“魔力甲基”(Magic Methyl)项。我们都知道,在分子某个特定位置加一个甲基,有时候会像变魔术一样,让活性飙升几十上百倍。这背后的原因往往很复杂,可能就是这个甲基精准地排开了一个关键的水分子,或者填补了一个疏水小坑。现在,Glide WS 也能像一个老道的化学家一样,识别出这种潜力。这标志着计算工具正在从单纯的物理打分,向融入化学直觉和经验的方向进化。
当然,老话还得说,垃圾进,垃圾出(garbage in, garbage out)。这一切的前提是你的蛋白结构要准备得足够好,质子化状态、氨基酸侧链构象等等都得处理妥当。好在 Schrödinger 也把这部分流程自动化了,算是尽可能减少了人为操作的失误。
Glide WS 并不是什么颠覆性的“银弹”,它解决不了药物研发的所有难题。但它确实把分子对接这个我们用了几十年的老工具,打磨得前所未有的锋利。对于所有依赖计算化学来驱动新药发现项目的团队和公司来说,这绝对是一个值得立刻评估和尝试的新武器。
📜Title: Glide WS: Methodology and Initial Assessment of Performance for Docking Accuracy and Virtual Screening
📜Paper: https://doi.org/10.26434/chemrxiv-2025-0s90h
2. AI 导航分子跃迁:告别盲人摸象式计算
做分子动力学模拟的研究者都明白,这项工作大部分时间是相当单调的。99.9% 的 CPU 时间,都投入在观察分子在稳定构型中自我娱乐地振动,犹如盯着一锅水等待其沸腾。真正令人兴奋的“沸腾”时刻——即化学反应、蛋白质折叠或药物解离等“稀有事件”,转瞬即逝。要捕捉到从 A 态到 B 态的完整路径,简直比大海捞针更为艰难。
过去我们怎么干?靠猜。
我们管这个猜出来的参数叫“集体变量”(Collective Variables, CVs)。你得凭着化学直觉,或者说“祖传经验”,去赌哪个或哪几个几何参数(比如原子间距离、二面角)是描述这个转变的关键。
猜对了,模拟效率大大提升;猜错了?恭喜你,浪费了几十万个核心时,得到一堆无用数据。就像让你在不看地图的情况下,只靠一个指南针在亚马逊雨林里找一条传说中的密道,运气成分太大了。
这篇论文的作者们不打算再玩这种猜谜游戏了。他们拿出的方案,可以说非常“釜底抽薪”。
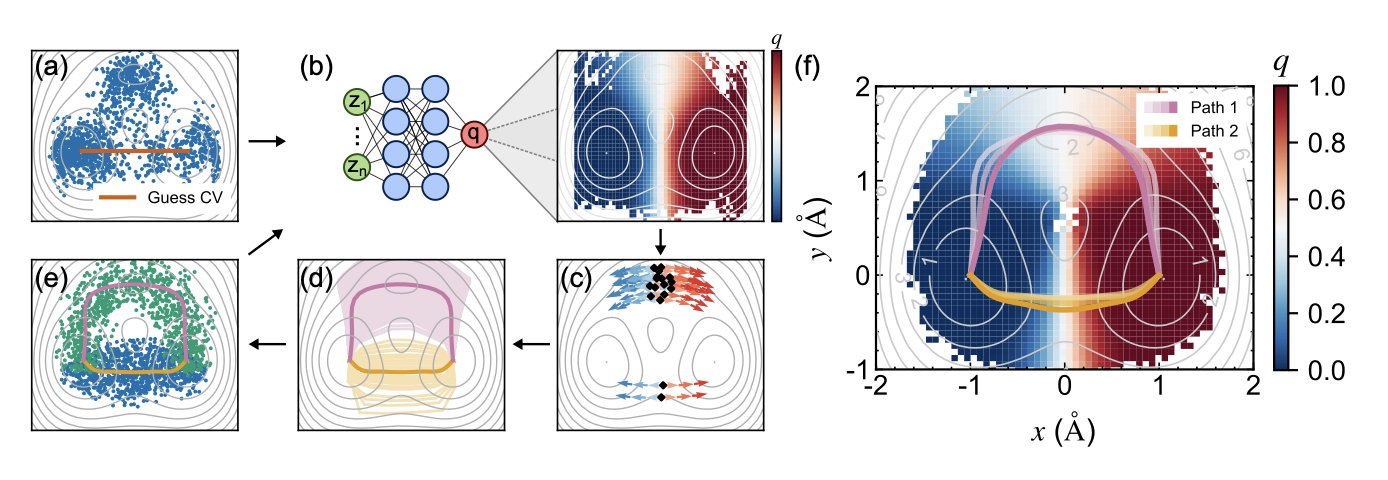
他们的核心思想是抓住一个叫“投递概率”(Committor Probability)的东西。这概念本身不新,而且理论上堪称完美的反应坐标。你可以把它想象成分子是个“信使”,要从 A 村庄送到 B 村庄。在路径上任何一个点,它有多大几率会先到 B 村庄,而不是滚回 A 村庄?这个概率就是 Committor。当这个概率是 50% 的时候,信使就正站在 A、B 两个村庄的分水岭上——也就是我们梦寐以求的过渡态。
问题来了,想知道这个概率,你就得先知道整个地形,这不又绕回去了吗?
作者用神经网络(NN)来打破这个死循环。他们不求一步到位,而是搞迭代。
第一步,先随便跑点模拟,数据糙点没关系,喂给神经网络,让它先学出一个粗糙的 Committor 函数模型。这就像派出了第一批侦察兵,画了张草图回来。
第二步,也是最关键的一步,是利用这张草图指导下一轮的模拟。他们把计算资源集中投放在 Committor 概率约等于 0.5 的区域,也就是那条“分水岭”附近。这就好比告诉第二批侦察兵:“别在村里瞎逛了,都去山脊上给我仔细搜!”这样一来,采样的效率指数级提高。
然后,重复这个过程。用新采集到的、更高质量的数据去训练一个更好的神经网络,得到一张更精确的地图,再用这张新地图去指导下一轮采样……如此循环往复,直到 Committor 函数收敛,不再有大的变化。这时候,你手里拿到的,就是一幅关于这条分子路径的高精度地图。论文里用 KL 散度的下降证明了这套方法的收敛性,很扎实。
更妙的是,他们还考虑到了现实中路径不止一条的复杂情况。当模拟跑出多条看起来不同的路径时,怎么判断它们是真正不同的“高速公路”,还是同一条路上的不同车道?他们引入了维诺单元(Voronoi cells)这个几何工具。简单说,就是根据路径点在空间中的归属,把那些本质上属于同一条“主干道”的岔路、小路都合并起来,避免了得到一堆乱七八糟的冗余结果。最终呈现给你的,就是几条干净、清晰、有代表性的核心路径。
他们拿这套方法在几个经典体系上跑了一遍,从二维模型到丙氨酸二肽构象翻转,再到经典的 Diels-Alder 反应,结果都相当漂亮。算出来的反应速率也跟之前的研究对得上。
这对做药的意味着什么?意味着我们能更有把握地去理解一个药物分子是怎么“扭”进靶点口袋的,或者它是怎么挣脱出来的——这对优化停留时间至关重要。这也意味着,在从头设计新分子时,我们可以更可靠地预测它可能的反应副产物。这套方法,把大量依赖“炼丹术士”直觉的工作,变成了一套有数据驱动、能自我修正的自动化流程。
他们也提到,如何从海量的候选参数中自动筛选出最有意义的 CVs,目前还只是个初步框架。但这已经是指明了一个非常有前途的方向。
📜Title: Following the Committor Flow: A Data-Driven Discovery of Transition Pathways
📜Paper: https://arxiv.org/abs/2507.21961
3. AI 生成分子进化,专攻硬核靶点
做基于结构的药物设计(SBDD),一线的人都清楚,最大的痛点是什么?数据。永远是数据。
高质量的蛋白 - 配体复合物三维结构,就那么些,PDB 数据库翻来覆去也就那些家底。相比之下,那些只有分子结构、没有靶点信息的配体数据,简直是汪洋大海。
大家都在想,怎么把这片海里的信息利用起来?
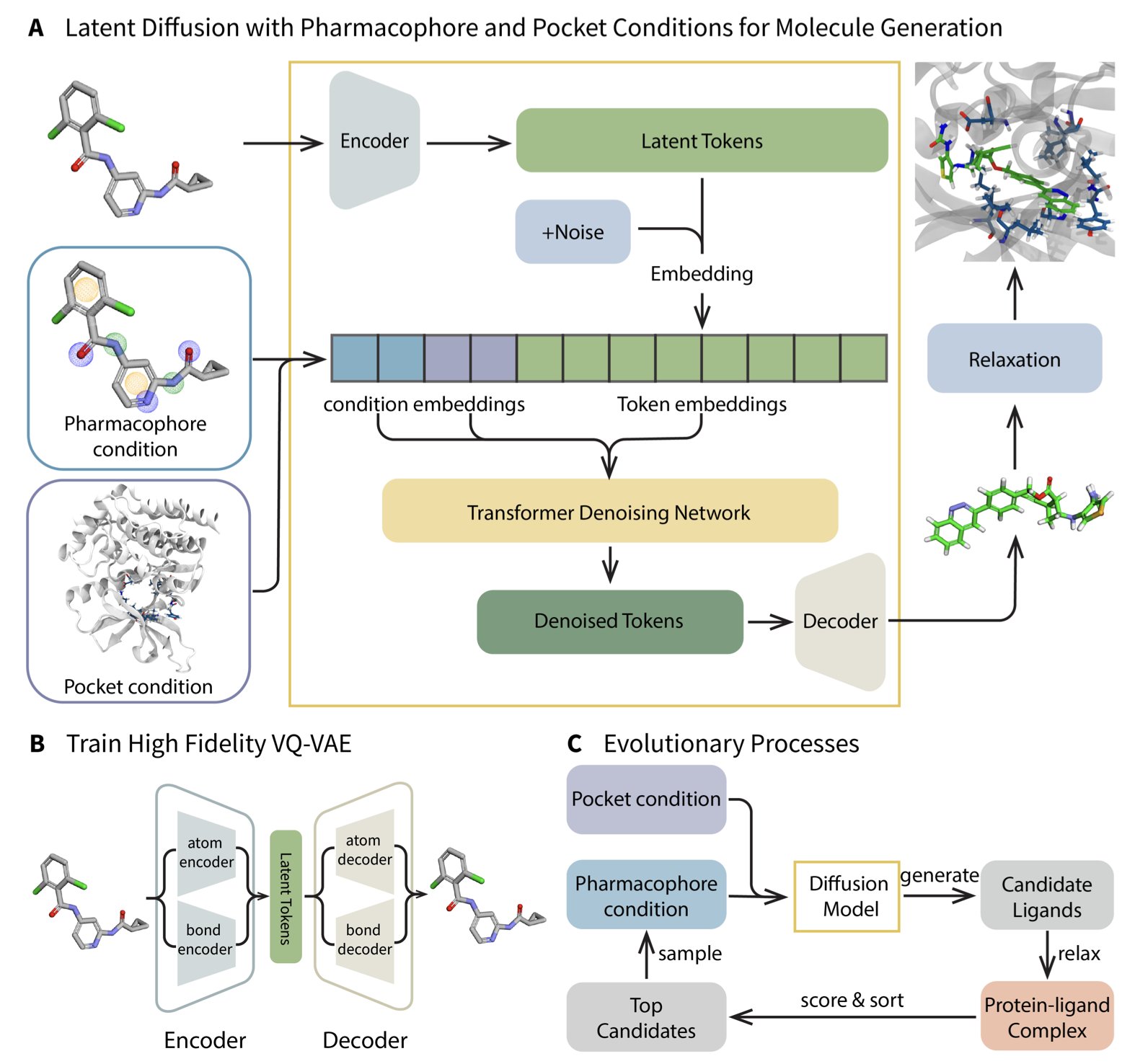
这篇 MEVO 的研究提供了一个相当精巧的解答。它并没有强行解决复合物数据不足的难题,而是切换了研究方向。其核心思路可以这样理解:我暂时不关注蛋白质口袋的具体特征,而是从庞大的分子库中,通过 VQ-VAE 和扩散模型,学习“有效分子”所应具备的“药效团骨架”。这就如同,我并不是通过观察“锁与钥匙”来制作钥匙,而是首先研究成千上万把能够打开各种锁的钥匙,以弄清楚“优秀钥匙”的通用设计原则。
这个想法本身不新鲜,但 MEVO 的高明之处在于它的后半段——进化优化。
AI 生成出来的分子,说白了,只是一些“看起来很美”的草图。它们符合药效团的统计规律,但能不能精准地嵌入目标蛋白那个刁钻的口袋里,那完全是另一回事。这时候,MEVO 的进化策略(Evolutionary Strategy)就登场了。它把这些生成的分子扔进蛋白口袋里,然后用基于物理的打分函数(比如对接能、力场计算)来当裁判,进行一轮轮“优胜劣汰”的筛选和变异。
这个过程就像是达尔文的自然选择,不过是在计算机里高速模拟。那些和口袋“八字不合”的分子被淘汰,而那些能形成关键相互作用的“天选之子”则被保留下来,并在此基础上继续“繁殖”和“变异”,迭代出亲和力更高的后代。
最关键的是,这个优化过程是“training-free”的,不需要复杂的模型再训练,可以直接用,还能轻松迁移到优化溶解度、渗透性等其他任务上。对于深受调参之苦的人来说,这简直是福音。
研究者们把 MEVO 扔去挑战了 KRASG12D 这个“老大难”靶点。做过 KRAS 的人都知道,它的结合口袋又浅又平,像个光滑的盘子,想让小分子牢牢站住脚非常困难。MEVO 不仅生成了全新的、具有高亲和力的分子骨架,而且通过自由能微扰(FEP)计算验证,其结合力与已知的强效抑制剂有一拼。这就很有说服力了。这表明 MEVO 不只是在制造符合 Lipinski 规则的“好看”分子,它生成的东西是真的有潜力成为先导化合物。
MEVO 是一个非常务实的工程解决方案。它把数据驱动的生成能力和物理规则驱动的优化能力漂亮地捏合在了一起。它承认了当前 AI 的局限,也发挥了经典计算方法的优势。这种融合,可能比追求某种单一技术的极致,更能解决我们药物研发中遇到的实际问题。
📜Title: Generative Molecule Evolution Using 3D Pharmacophore for Efficient Structure-Based Drug Design
📜Paper: https://arxiv.org/abs/2507.20130
4. ChemDFM-R:教 AI 像化学家一样思考
大部分所谓的“化学大模型”,本质上还是个语言学专家,而不是化学家。它们就像一个背了整本字典但不会造句的学生,能告诉你某个反应的名字,却根本搞不懂电子到底是怎么跑的,原子是怎么重新排布的。
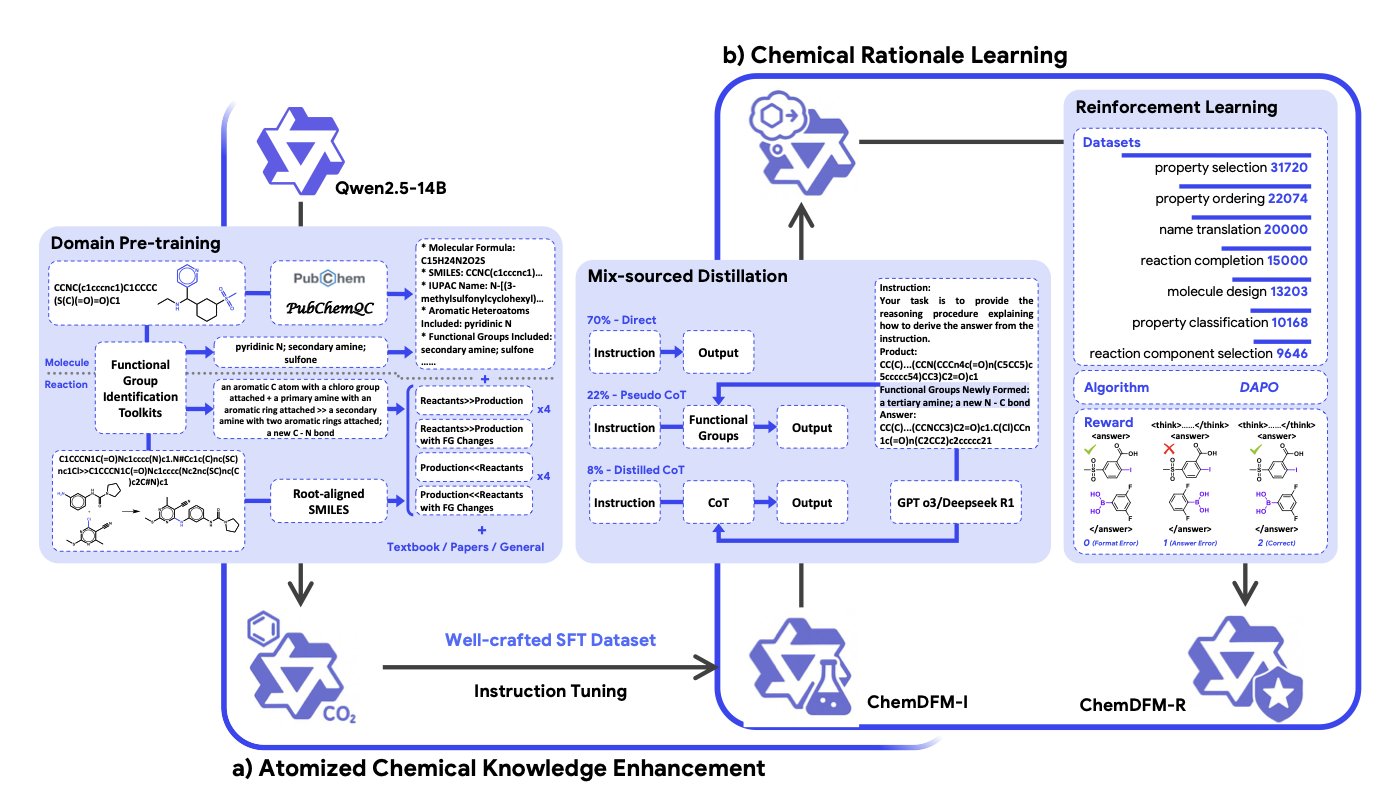
ChemDFM-R 干了一件非常“化学”的事。他们没有让模型去硬啃整个复杂的化学反应,而是把知识打碎,“原子化”到化学世界最基本的单元——官能团。
对于化学家来说,这就是思考的起点。看到一个分子,他们眼里不是一堆杂乱的碳氢氧氮,而是酯基、是羰基、是氨基……是这些决定分子“性格”和“行为”的关键部件。
论文的作者们显然深谙此道。他们教会了模型去识别分子里的官能团,以及这些官能团在反应中如何变化。
就像教一个孩子玩乐高。你不是直接给他看一个拼好的千年隼,而是先教他认识每一块积木——这个是 2x4 的砖,那个是带铰链的,它们各自有什么用,可以怎么拼在一起。当他理解了每块积木的“官能”,他才能真正开始创造,而不是死记硬背模型图纸。
ChemDFM-R 的训练方法也相当聪明。
它用了一种“混合来源蒸馏”的策略。一方面,用专家们整理好的、精确的化学知识(就像教科书里的金科玉律)来给模型打下坚实的基础。另一方面,它又去向那些更强大的通用大模型(比如 GPT-4)学习通用的推理和逻辑能力。就像一个学生,既有专业课老师的悉心指导,又能旁听哲学系的逻辑课。
最后,再用上强化学习这个“严师”,不断对模型的化学“答卷”进行打分和纠错,直到它真正“开窍”。
所以结果是什么?
它不只是在各种化学基准测试中得分很高还有它的可解释性。
当 ChemDFM-R 预测一个反应时,它不只是给你一个产物。它会像个真正的化学家一样,一步步告诉你它的思考过程:“因为底物中存在一个酯基,而反应体系中有这个亲核试剂,所以亲核加成 - 消除反应会在这里发生……”
它把那个黑箱子撬开了一道缝,让你能看到里面的齿轮是怎么转动的。对于做药物研发的人来说,这种可解释性比单纯 99% 的准确率要有价值得多。我们必须知道为什么,才能信任它的预测,才能基于它的建议去设计下一步的、可能耗资数百万美元的实验。否则,它和算命先生有什么区别?
当然,这一切的背后,是研究者们堆出的一个“数据金矿”——ChemFG 数据集。他们处理了 1200 万篇文献、3000 万个分子和 700 万个化学反应,把这些信息都提炼成了模型能理解的、以官能团为核心的知识。这手笔,堪称是为 AI 建了一座化学领域的国会图书馆。
ChemDFM-R 像一个知识渊博但经验尚浅的博士生。你可以和它讨论,可以质疑它的逻辑,也可以从它的思路里获得启发。
📜Paper: https://arxiv.org/abs/2507.21990v1
5. LLM 生成合成健康数据:高效但离不开人
在药物研发和健康科技领域,谁都知道搞到高质量的真实世界患者数据有多头疼。HIPAA、隐私、伦理,条条框框能把人逼疯。
所以,我们常常把目光投向合成数据。Synthea 是个不错的开源工具,它能生成虚拟患者记录,但问题是,要为一种新疾病从零开始构建一个模块,那过程简直是手工作坊式的,又慢又乏味。
现在,MITRE 的研究者把 LLM 这个时髦的工具扔进了这个老问题里。他们的想法可不是简单粗暴地对 AI 说:“喂,给我写个 2 型糖尿病的模块。”那纯属自找麻烦,等着 AI 给你创造出医学奇迹。
他们建立了一套四阶段“流水线”:
- 喂给它蓝图: 首先,你得给 LLM 一份扎实的、权威的临床指南。记住,垃圾进,垃圾出,这个古老的法则在 AI 时代依然有效。
- 拿到初稿: LLM 会吐出一个初始的 Synthea 模块。把它想象成一块刚从采石场运出来的、未经雕琢的大理石。
- 现实检验(Sanity Check): 这就是人类专家登场的时候了。检查两件事:代码语法对吗?(程序能跑起来吗?)以及,更要命的,这在临床上说得通吗?(疾病进展、治疗方案,符合医学逻辑吗,还是 AI 又在自由发挥了?)
- 迭代,迭代,再迭代: 你把发现的错误反馈给 LLM,“这部分不对,改”,然后重复这个过程。研究者管这叫“渐进式优化”,我更愿意称之为“训练一个绝顶聪明但偶尔会犯傻的实习生”。
结果相当不错。他们声称最终模块的临床准确性“接近完美”。但我们都懂,“接近完美”这个词背后,是人类专家在不厌其烦地揪出 LLM 那些防不胜防的“幻觉”。要是没人把关,你很可能会得到一个被 AI 建议用棒棒糖治疗高血压的虚拟病人。
所以,这会是未来吗?
对于从临床指南这种结构化信息源生成结构化数据的任务来说,答案是肯定的。它能把过去需要一周的手动编码工作,压缩成可能一天就能完成的审查和优化。这是个巨大的效率提升。但这绝不是那个能让你“解雇数据科学家”的魔法按钮。
这套方法的真正价值,不在于 LLM 生成的初稿,而在于它建立了一个流程。这个流程把 LLM 当成一个强大的工具,同时又把人类的专业知识置于最终裁决者的位置。工作的瓶颈,从繁琐的编码,转移到了高阶的、需要深厚功底的专家验证上。
📜Title: Leveraging Generative AI to Enhance Synthea Module Development
📜Paper: https://arxiv.org/abs/2507.21123v1