
目录
- 通过在图神经网络中加入基于 3D 优化结构的精细化学特征,DILIGeNN 模型在预测药物性肝损伤(DILI)方面取得了当前最优的性能。
- GHCDTI 通过融合图小波变换和对比学习,不仅提升了药物 - 靶点相互作用预测的准确性,还首次在模型中考虑了蛋白质的动态柔性。
- 默沙东的这项研究证明,将高质量的公共数据与公司内部数据巧妙结合,尤其是通过多任务学习,可以显著提升 ADME 预测模型的准确性和泛化能力。
1. DILIGeNN:用更精细的 3D 特征预测肝损伤
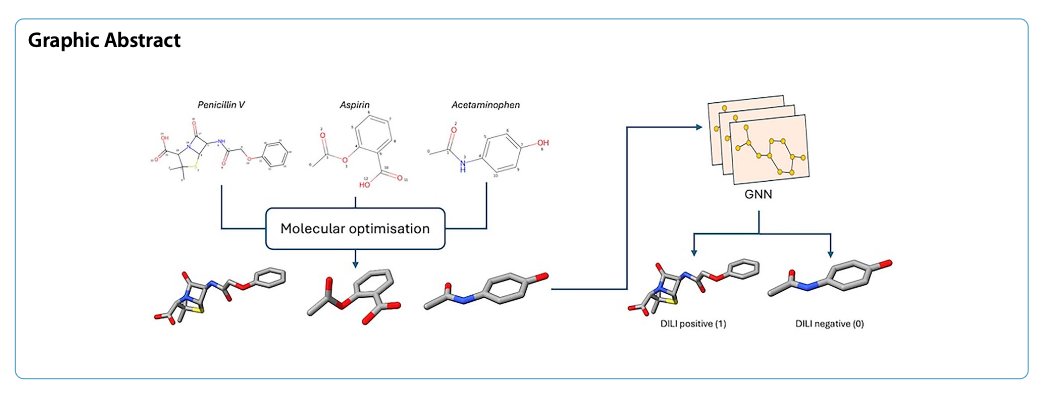
药物性肝损伤(DILI)是药物研发中令人头疼的「拦路虎」。它常常在临床试验后期甚至药物上市后才暴露出来,导致经济损失和严重的公共健康问题。如果我们能在药物研发的早期,就用 AI 模型把那些有 DILI 风险的分子给筛出来,那将是巨大的提升。
现在的图神经网络(GNN)在预测分子性质方面很流行。但大多数 GNN 模型,吃的「原料」都比较粗糙。它们通常只看分子的 2D 连接关系,也就是哪个原子和哪个原子连在一起。这就好比看一张简笔画,虽然能看出大概轮廓,但丢失了很多细节。
DILIGeNN 的创新:从「简笔画」到「工笔画」
这篇论文的 DILIGeNN 模型,就是想给 GNN 喂更精细的「工笔画」。
它的思路是:在把一个分子喂给 GNN 之前,先对它进行一次「精加工」。
1. 3D 结构优化 :研究者们首先用计算化学的方法,为每个分子生成一个能量最低、最接近其真实状态的 3D 构象。
2. 提取精细特征 :然后,他们从这个优化后的 3D 结构里,提取出了一系列传统 GNN 模型不会用到的信息,比如:
* 更精确的键长 :双键就是比单键短,这个几何信息现在被明确地告诉了模型。
* 部分电荷 :分子中哪个原子带正电荷,哪个带负电荷,电荷分布情况如何。
* 原子轨道杂化类型等。
把这些更丰富、更符合化学现实的特征,作为图的节点和边的属性,再输入到 GNN 中去训练。这就相当于,我们给模型的「原料」里,加入了更多有价值的信息。
效果如何?
结果证明,吃「精加工」原料的模型,确实比吃「粗粮」的模型更聪明。
在最关键的 FDA DILI 数据集上,DILIGeNN 的 AUC 达到了 0.897,这是一个非常高的水平,超过了以往报道的其他模型。
这种方法的优势并不仅仅局限于 DILI 预测。研究者们把同样的方法,用在了其他几个公开的基准数据集上,包括:
在所有这些任务上,DILIGeNN 都取得了当前最优或接近最优的性能。这证明了,提供更精细的化学特征,是一种具有普适性的、能够提升 GNN 模型性能的有效策略。
在 AI 药物发现领域,我们不应该仅仅满足于算法层面的创新。回归到化学本身,思考如何为模型提供更高质量、更具信息量的输入,同样是提升模型性能的关键。
DILIGeNN 仅仅依赖于分子的化学结构特征,就能做出如此准确的预测,这对于在药物发现早期阶段快速筛选和评估候选化合物,非常有价值。
📜Paper: https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01068-3
💻Code: https://github.com/tlee23-ic/GNN_DILI
2. GHCDTI:一个更懂蛋白动态的 DTI 预测模型
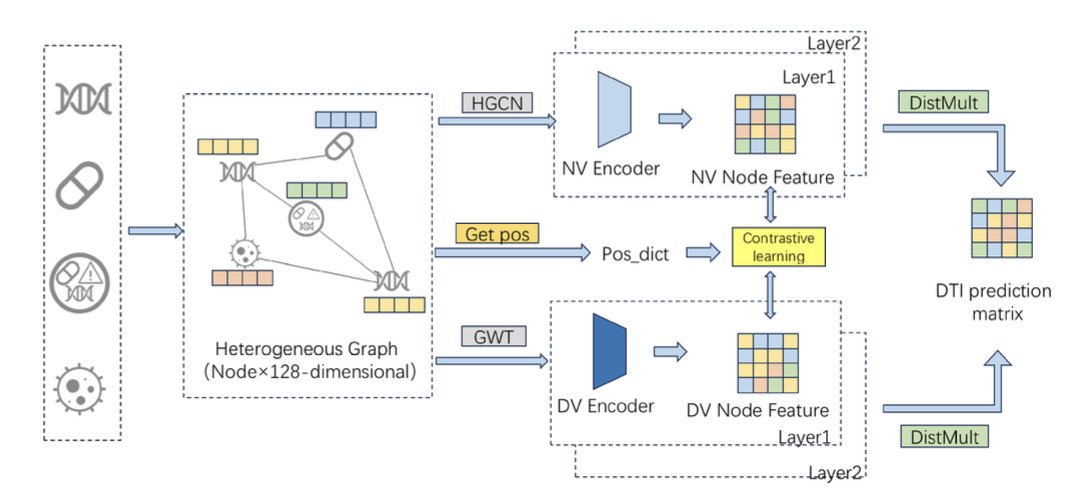
预测一个新的小分子会和人体内哪个蛋白质靶点结合(DTI 预测),是药物发现的起点。现在的 AI 模型,尤其是图神经网络,已经能在这方面做得不错。但它们大多都有几个共同的软肋:
- 把蛋白质当成刚体 :很多模型只看蛋白质的静态三维结构,但我们都知道,蛋白质在体内是柔软的、会动的。它的构象变化,往往是和药物结合的关键。
- 数据不平衡 :已知的药物 - 靶点结合数据,相对于茫茫的化学和蛋白空间,只是沧海一粟。正样本(会结合)极少,负样本(不会结合)海量。这会让模型倾向于「躺平」,直接猜「不结合」,准确率还挺高,但对我们没用。
- 黑箱问题 :模型说这个分子能结合,但为什么能结合?哪个部分起了关键作用?很多模型答不上来。
这篇论文的 GHCDTI 模型,就试图一次性解决这三个难题。
怎么让模型看到「动态」的蛋白质?
GHCDTI 用了一种叫「图小波变换」的数学工具。
你可以这么理解:一张图片,我们可以把它分解成高频信号(轮廓、细节)和低频信号(背景、色块)。同样,一个蛋白质的结构图,也可以通过图小波变换,分解成不同的「频率」成分。
GHCDTI 通过同时学习这两个部分的信息,就得到了一个更全面的蛋白质表征。它既知道这个蛋白长什么样,也知道它哪些地方喜欢「动」。这对于理解药物如何与动态的结合口袋相互作用,至关重要。
用对比学习对抗数据不平衡
为了不让模型在数据不平衡问题上「躺平」,GHCDTI 用了「对比学习」的策略。
它为每个节点(药物或蛋白)生成了两个不同的「视图」:一个是从拓扑结构来的,一个是从上面说的小波变换来的。然后,它强制模型去学习,让同一个节点在这两个不同视图里的表示尽可能地相似,而与其他节点的表示尽可能地不同。
这就好比,我们让一个学生同时看一篇文章的原文和译文,然后要求他能认出这两篇文章讲的是同一个故事。通过这种方式,模型被迫去学习更本质、更鲁棒的特征,而不是仅仅记住一些表面现象。
融合多源信息,提升可解释性
GHCDTI 还把药物的 2D 结构、蛋白质的 3D 结构以及已知的生物活性数据,通过一个跨图注意力机制融合在一起。这个注意力机制,就像给模型配了一副「透视眼镜」,它能告诉我们,在做预测时,模型主要「看」了药物的哪个基团和蛋白质的哪个区域。这就为我们理解和优化分子提供了直接的线索。
从结果来看,GHCDTI 在多个基准数据集上都取得了当前最优的性能,而且在「冷启动」(预测全新的药物或靶点)场景下表现也很好。
📜Paper: https://www.nature.com/articles/s41598-025-16098-y
3. 默沙东经验:公共 + 私有数据可改善 ADME 预测
每个大型药企(Big Pharma)都坐拥一个金矿——几十年来积累的内部化合物和 ADME(吸收、分布、代谢、排泄)数据。用这些数据来训练 AI 模型预测新分子的 ADME 性质,是研发人员日常工作的一部分。
但我们一直面临一个问题:我们用自己家数据训练出来的模型,去预测一个化学结构和我们以往项目完全不同的外部化合物,还准吗?反过来,一个用公共数据库训练的模型,能准确预测我们内部那些结构独特的在研分子吗?
答案往往是:不一定。
模型很可能会「偏科」,只对自己熟悉的数据类型表现良好。这就是所谓的「适用域」(Applicability Domain)问题。
默沙东的这篇研究,就对这个问题做了一次非常系统和坦诚的探讨。
怎么解决模型的「偏科」问题?
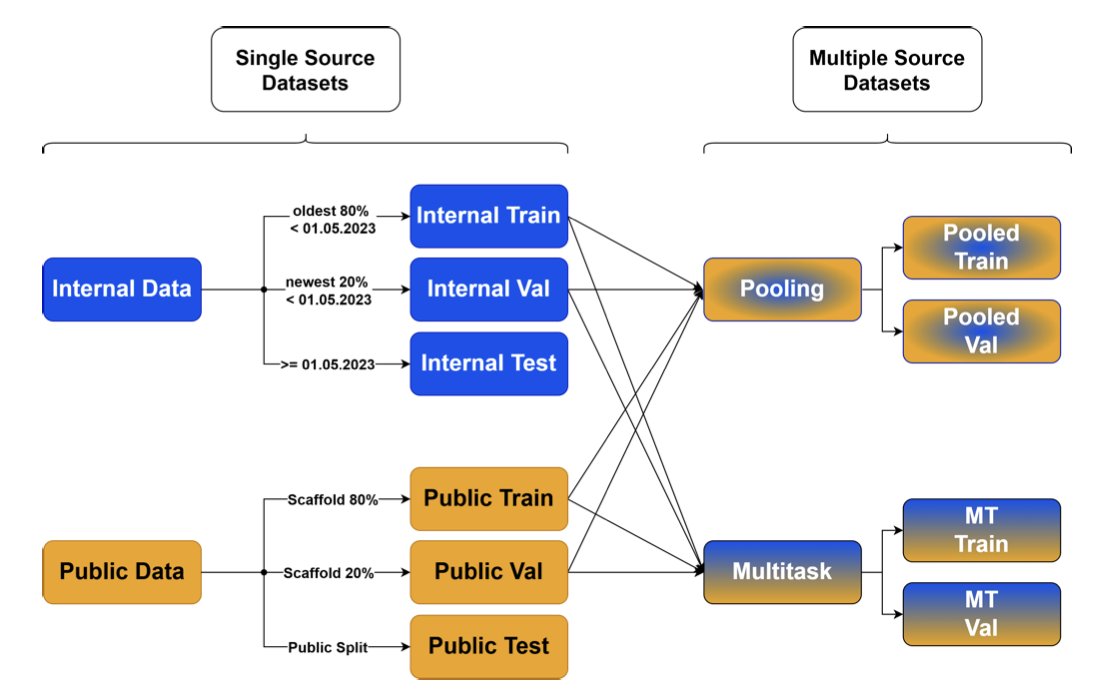
他们的思路是:既然只吃一种食谱会营养不良,那就让模型「兼收并蓄」,把公共数据和公司内部数据这两盘「大餐」都给它吃。
但怎么「吃」是有讲究的。
1. 简单混合(Pooled Single-Task) :最简单的办法,就是把两个数据集直接倒在一起,训练一个模型。这样做有时候管用,但如果两个数据集的分布差异很大,或者数据质量参差不齐,可能会适得其反,把模型「带偏」。
2. 多任务学习(Multi-Task Learning) :这是这项研究中表现最好的方法。你可以把它想象成,我们让一个学生同时学习两个既有联系又有区别的科目,比如物理和化学。在学习的过程中,他会发现一些底层的通用规律(比如能量守恒),这些规律在两个科目里都适用。
多任务学习就是这样。模型在底层共享一部分网络参数,用来学习所有数据共有的化学规律。而在上层,它又为每个数据集(公共和私有)保留了各自独立的「专家」模块,用来学习各自独特的 SAR。通过这种方式,模型既学到了「共性」,又保留了「个性」,最终在预测两个数据集时都能表现得更好。
数据说了算
默沙东的研究人员用他们内部的海量数据,结合像 Biogen 报告里公开的高质量数据集,对六个关键的药代动力学终点(比如清除率、生物利用度等)进行了建模。
结果在几乎所有的测试中,使用混合数据训练的模型,特别是多任务模型,其性能都优于只使用单一来源数据训练的模型。无论是在预测内部测试集,还是在预测外部公共测试集上,准确率和可靠性都有了实打实的提升。
所以第一,不要固步自封。即使是像默沙东这样拥有海量高质量内部数据的公司,积极拥抱和整合外部公共数据,依然能带来巨大的价值。这扩大了模型的视野,让它能更好地应对未来项目中可能出现的全新化学骨架。
第二,多任务学习是处理异源数据的一个强大工具。它提供了一种更优雅的方式来融合信息,而不是简单粗暴地把数据堆在一起。
📜Paper: https://doi.org/10.26434/chemrxiv-2025-7sbr0