
目录
- PARSE 聚焦于蛋白质上保守的局部微环境,而非整体结构,从而更精准地预测功能和活性位点,尤其擅长研究「黑暗蛋白质组」。
- Honeybee 框架整合了癌症的各类数据。研究发现,简单的临床文本,其预测能力超过了复杂的基因组或影像数据。
- Boltzina 让一个快速但粗略的工具完成初步工作,再由一个精准但缓慢的 AI 做最终评估,在药物虚拟筛选中兼顾了速度与准确性。
1. AI 预测蛋白功能:看局部,不看全局
AlphaFold 绘制出数亿蛋白质的结构图谱,如同给了我们一张宇宙地图。但地图上绝大多数「星球」(蛋白质)的功能仍然未知,它们是广阔的「黑暗蛋白质组」。我们虽有其三维结构,却不知其功能。
以往预测蛋白质功能,主要依靠 BLAST 比较氨基酸序列,或 Foldseek 比较整体三维结构。这两种工具都基于整体相似性。若两个蛋白质的序列或整体折叠差异较大,它们就会被判定为无关联。这种方法好比通过面部相貌判断亲缘关系,可能会忽略掉那些外貌不同但拥有共同家族特征的远亲。
PNAS 上的一篇论文提出,AI 需要一副能识别这些局部「家族特征」的放大镜。
识别功能的「指纹」
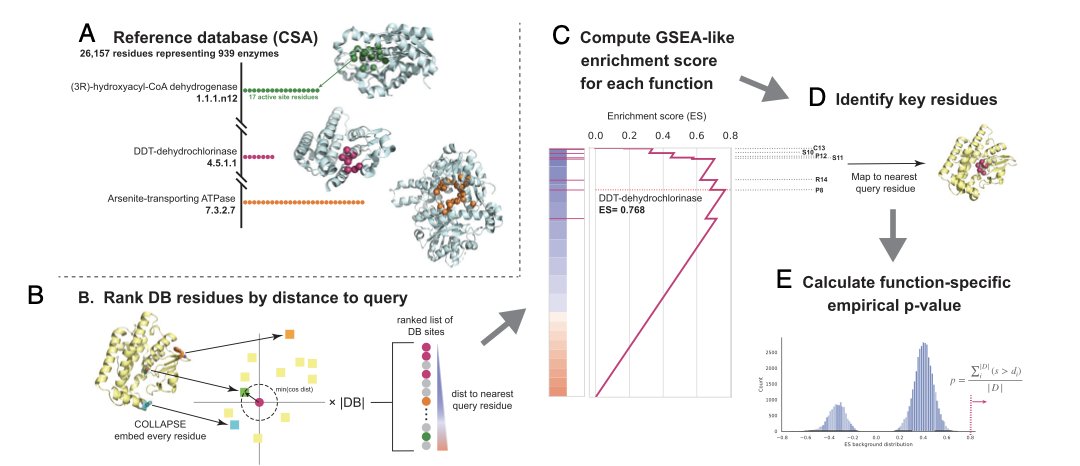
新方法 PARSE 基于一个生物学直觉:决定蛋白质功能的,是其微小且在进化中高度保守的活性或功能位点,而非庞大易变的整体结构。
这个位点就是蛋白质的「指纹」。PARSE 的工作流程跳过了对整体结构的比较:
首先,它将蛋白质分解为无数个重叠的局部微环境,每个环境都以一个氨基酸为中心。
接着,它使用 COLLAPSE 工具,将每个局部环境的三维几何信息,转换为一个数字化的「指纹」(嵌入)。
最后,它在一个大型数据库中,直接比较不同蛋白质的局部「指纹」,寻找高度相似的区域。
精准定位功能位点
通过识别「指纹」,PARSE 的预测能力大幅提升。在预测蛋白质整体功能方面,它的表现与复杂的深度学习模型相当,F1 分数超过 85%。
PARSE 的优势在于精准定位。它在预测「一个蛋白是激酶」的同时,还能高置信度地指出构成其活性位点的具体氨基酸。其定位精度超过了 DeepFRI 等现有方法。
这种精确的靶点信息对药物研发至关重要。研发人员需要的是精确的「靶点地图」来指导定点突变或药物设计,而非模糊的功能标签。
该方法速度很快,足以扫描整个人类蛋白质组。研究者利用这一优势,在一些功能未知的蛋白质中发现了新的潜在功能位点。
这项工作为如何构建更符合生物学原理的 AI 提供了范例。它表明,研究关键的局部特征,有时比分析整体结构更有效。
📜Title: Protein functional site annotation using local structure embeddings
📜Paper: https://www.pnas.org/doi/10.1073/pnas.2513219122
2. AI 肿瘤研究:临床数据才是王道
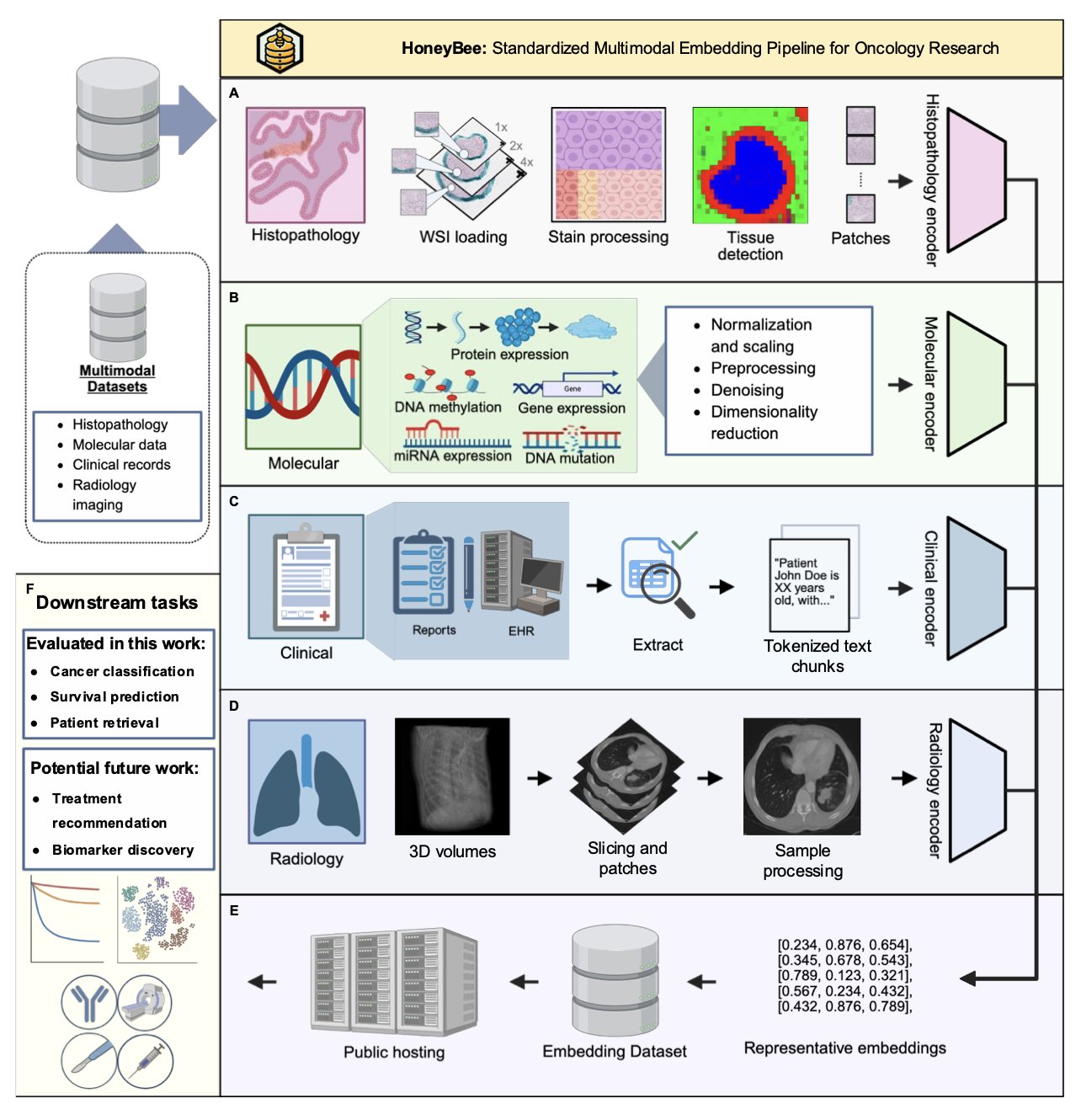
肿瘤研究领域的数据量巨大,但开发其价值却充满挑战。一个癌症病人会产生多种数据:医生的临床笔记、病理科的全切片扫描(Whole Slide Imaging, WSI)、放射科的 CT 或 MRI 影像,以及基因测序图谱。这些数据分散在不同部门,如同语言不通的专家,难以描绘出患者的全貌。
业界期待一个「总翻译」来整合所有信息,提供一个综合的患者视图。Honeybee 框架正是实现这一目标的尝试。
Honeybee 如何拼凑完整拼图
Honeybee 是一个开源框架,它能处理临床文本、病理影像、放射影像和分子数据。通过一系列 AI 基础模型,它为每位患者生成一个统一的「数字指纹」(embedding)。
这个「数字指纹」有多种用途,例如预测生存期、进行疾病分类,或是在数据库中快速检索相似病例。
整合所有数据,直觉上能带来更好的结果。但研究者在一万多名 TCGA 患者数据上进行严格测试后,得到了一个出乎意料的发现。
最强预测者:临床文本
在比较不同数据类型的独立预测能力时,胜出的是临床数据,而非技术上更复杂的全切片扫描或基因组数据。
由医生记录的临床文本,在 33 种癌症分类任务中,仅凭其生成的「数字指纹」,准确率就达到 98.5%。在检索相似病人方面,该数据的表现同样最佳。
这好比预测一辆车的性能,人们可能以为需要发动机设计图(基因组)和风洞测试数据(影像),结果最准的预测却来自记录了每次加油和维修的行车日志。
这个结果表明,在 AI 时代,高质量结构化临床数据的价值可能被低估了。
当然,多模态数据依然有其价值。研究者发现,在预测总生存期这类更复杂的问题时,整合所有数据能取得比单一临床数据更好的结果。这表明,不同数据在解决不同问题时各有优势。
另一个意外:通才胜过专家
研究者还测试了哪种 AI 模型最擅长解读临床笔记。直觉上,专门用医学文献训练的医疗 AI 应有优势。
但测试结果显示,一个通用的模型(Qwen3)表现超过了专门的医疗模型。
这可能是因为临床记录多为描述性自然语言,而非高度专业化的术语,而处理自然语言正是通用大语言模型的长项。
Honeybee 框架作为一个开源工具,能够整合多种癌症数据。这项研究也用数据得出了两个重要启示:
第一,临床数据拥有巨大的潜力,其价值不应被忽视。
第二,在选择 AI 工具时,通用模型在某些任务上可能优于专用模型。
📜Title: Honeybee: Enabling Scalable Multimodal AI in Oncology Through Foundation Model–Driven Embeddings
📜Paper: https://arxiv.org/abs/2405.07460
3. Boltzina:让 AI 筛选药物既快又准
虚拟筛选长期存在一个矛盾:速度与准确性难以兼得。
一方面,传统工具如 AutoDock Vina 是「快枪手」,能快速处理百万级分子,但其假阳性率高,不够精准。
另一方面,新兴的 AI 模型如 Boltz-2 是「神射手」,预测精准。但它计算过程耗时,若用于筛选百万级分子库,项目周期将无法接受。
研究人员一直寻求兼具速度和准确性的工具。Boltzina 为此提供了一个务实的解决方案。
快枪手与神射手联手
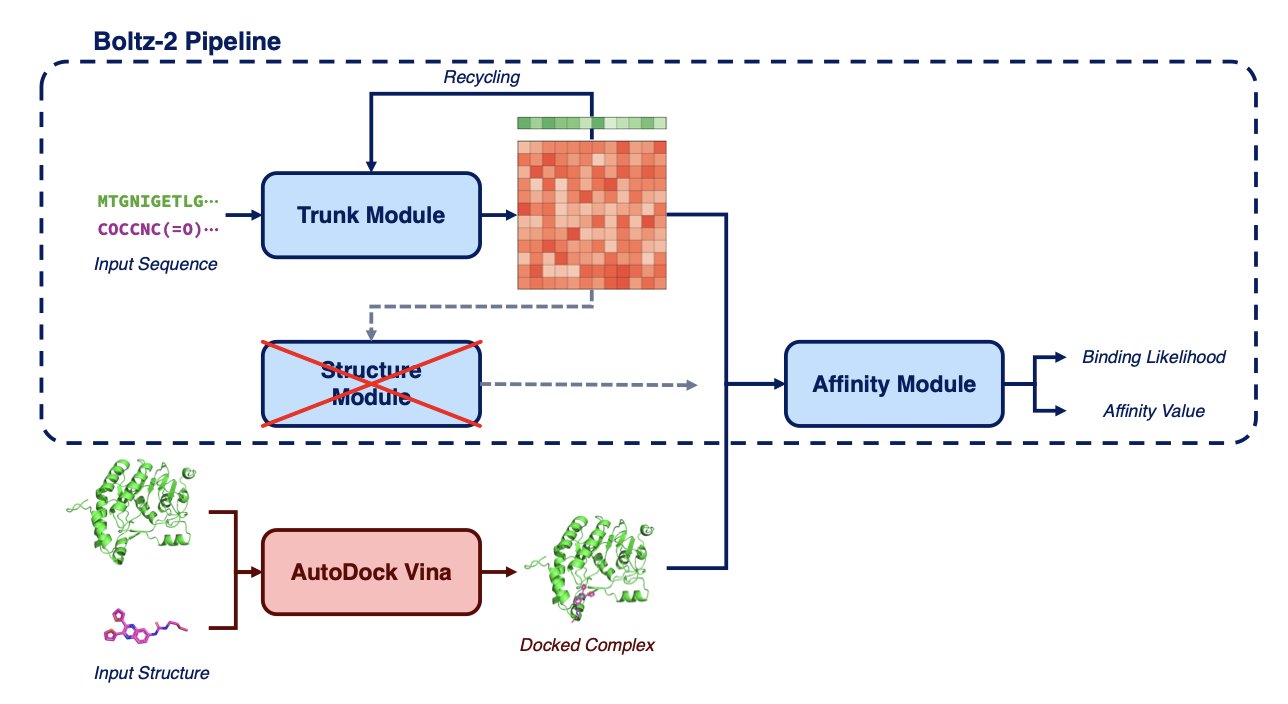
Boltzina 的思路是流程再造,让两种工具分工合作。
流程如下:
第一步,由「快枪手」Vina 上场,将大量分子与蛋白质靶点进行对接,生成所有可能的对接「构象」(pose)。这一步追求的是速度和覆盖面,而非精准度。
第二步,由「神射手」Boltz-2 进行最终评估。Boltzina 将 Vina 生成的构象直接提交给 Boltz-2 的打分模块,由后者评估每个构象的可靠性并打分。
Boltzina 省略了 Boltz-2 最强大也最耗时的「结构共折叠」步骤,只利用其高效的打分模块。这好比请艺术鉴赏家为一批现成作品估价,而无需他从头雕刻。
组合策略的效果
该流程在保持 Boltz-2 级别高筛选富集率的同时,将速度提升了近 12 倍。过去需要一周的计算量,现在一天内即可完成。
研究者还进行了一些优化。他们发现,对 Vina 生成的多个构象的 Boltzina 得分进行平均,可以得到更稳定可靠的结果。
他们还提出一个两阶段筛选策略:先用 Boltzina 从大型化合物库中快速筛选出数千个候选分子,再用完整的 Boltz-2 对这些候选分子进行精选。对于资源有限的项目,这是一个平衡了成本与收益的合理工作流。
Boltzina 是一个工程解决方案。它并未发明一个全新的工具,而是将两个各有优劣的现有工具组合,解决了一线研发人员面临的实际问题。
它让 Boltz-2 级别的预测能力,首次可以被用于几十万到几百万规模化合物库的实际筛选项目。
📜Title: Boltzina: Efficient and Accurate Virtual Screening via Docking-Guided Binding Prediction with Boltz-2
📜Paper: https://arxiv.org/abs/2508.17555v1
💻Code: https://github.com/ohuelab/boltzina