
目录
- ChemGenXplore 将多种复杂的化学基因组学分析功能,打包成一个对实验科学家极其友好的交互式网络平台。
- Chem3DLLM 通过一种巧妙的可逆压缩编码,成功地将 3D 分子结构「翻译」成 LLM 能理解的文本,为基于结构的药物设计开辟了一条全新的、端到端的解决路径。
- AuroBind 真正将结构预测与大规模生物活性数据对齐,并在无结构信息的孤儿靶点上找到了皮摩尔级活性分子,这才是虚拟筛选该有的样子。
1. ChemGenXplore:化学基因组学数据探索新工具
做高通量筛选的都懂,拿到数据只是第一步。真正头疼的是面对那堆积如山、维度复杂的数据。
一个化学基因组学筛选下来,成千上万个基因和几十个处理条件,数据矩阵大得吓人。通常,这些数据得先交给生物信息专家处理,一来一回,从产生数据到获得洞见,中间总有不小的延迟。
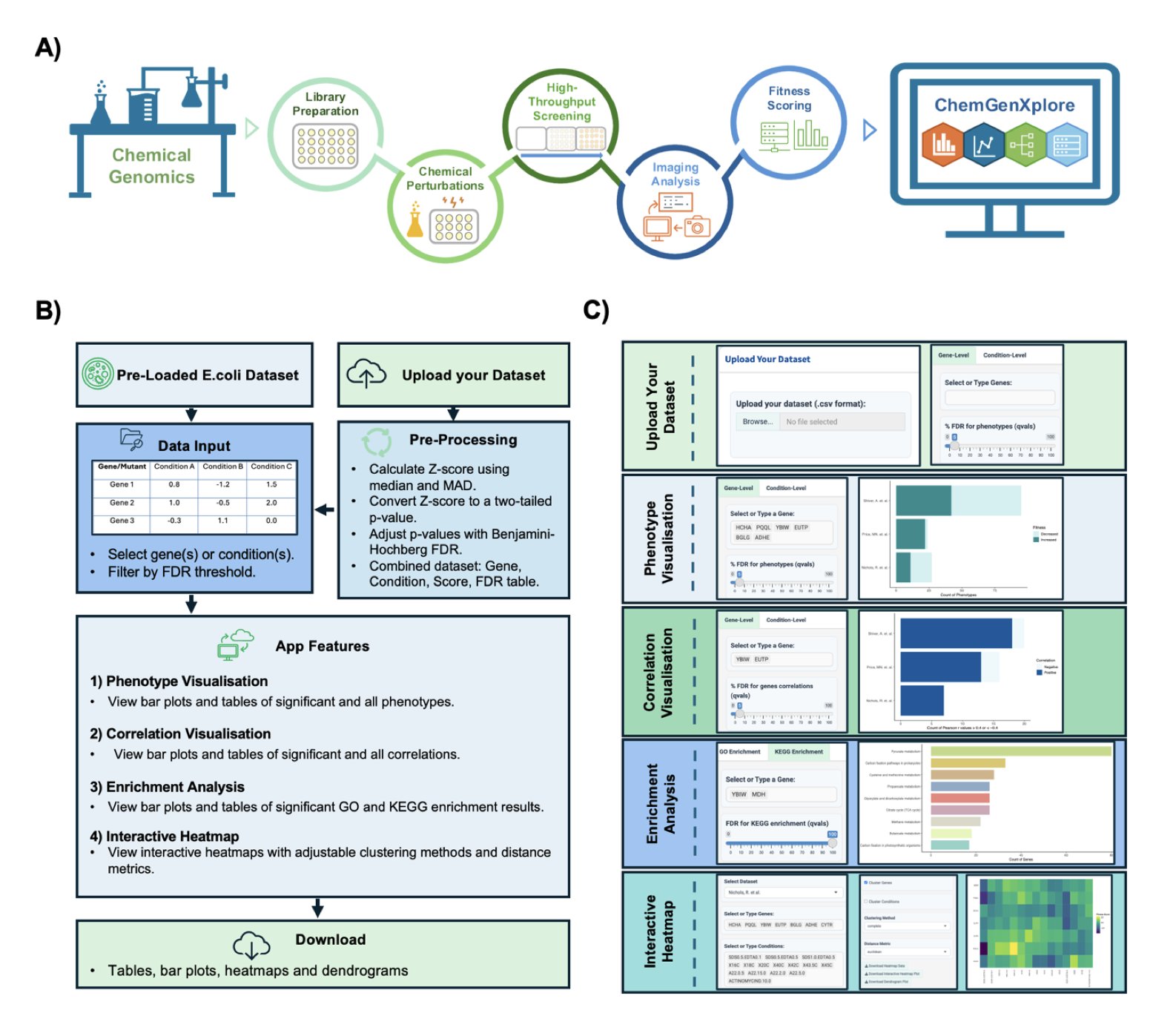
现在,研究者们开发了一个叫 ChemGenXplore 的工具,这东西本质上是一个 Shiny 应用。对于熟悉 R 语言的人来说,Shiny 能把分析脚本变成一个交互式的网页,让不懂代码的人也能直接上手。这正是 ChemGenXplore 的核心价值:它把一套标准的化学基因组学分析流程,封装进了一个任何人都能点击操作的图形界面里。
我们来看看它具体能做什么。
首先是表型可视化。假设你用一个化合物库处理了一个酵母基因敲除文库,想快速知道哪些基因的缺失会让酵母对你的某个「明星分子」特别敏感。过去,你可能要在表格里挣扎。现在,直接在 ChemGenXplore 里输入基因名或处理条件,一个交互式的条形图就跳出来了,还能根据 FDR(错误发现率)阈值进行筛选,保证你看到的是统计上靠谱的结果。这极大地加速了初步「hit」的筛选。
接下来是相关性分析,这是挖掘生物学故事的关键。如果两个基因在所有化合物处理下的表型谱(fitness profile)非常相似,那它们很可能在同一条生物学通路里干活。反过来,如果两个化合物在整个基因文库上产生的「杀伤谱」类似,它们的作用机制或许也相近。这个工具能帮你计算这些相关性,并用图表清晰地展示出来。设定一个比如 ±0.4 的相关性阈值,就能快速锁定那些值得深入研究的基因或化合物组合,避免在噪音数据里大海捞针。
然后是功能富集分析。找到了几十个敏感基因,然后呢?它们是干什么的?ChemGenXplore 集成了 GO 和 KEGG 富集分析。你把基因列表扔进去,它会自动告诉你这些基因是不是扎堆出现在某些特定通路里,比如 DNA 损伤修复、线粒体呼吸链或者某个代谢途径。对于药物研发来说,这就是在直接给出关于化合物作用机制的线索,是假设生成(hypothesis generation)的利器。
最后,交互式热图。热图是展示这类高维数据的经典方法,能让你一眼看清全局。哪些基因对哪一类化合物特别敏感,数据里的聚类模式是什么样的,一目了然。这个工具里的热图不仅能自定义聚类算法,还是交互式的,你可以放大、平移,鼠标悬停查看具体数值。这比看一张静态的图片要强大太多了。
ChemGenXplore 的特点在于「集成」和「易用」。它把生物信息学家的命令行工具,变成了一个实验科学家在浏览器里就能把玩的「玩具」。这大大缩短了从数据到洞见的距离,让做实验的人能亲自探索自己的数据,快速验证想法。。
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.13.670066v1
2. LLM 终于能「看懂」3D 分子了?Chem3DLLM 技术解析
谁能不希望有个工具能直接跟它说:「嘿,这是我的靶点口袋,给我设计个高活性的分子出来」,然后它就能给你一个靠谱的 3D 结构?我们一直在朝这个方向努力,但语言模型(LLM)的出现让事情变得有点尴尬。LLM 处理文本是把好手,可分子,尤其是它的 3D 构象,根本不是一回事。你没法直接把一堆原子坐标喂给一个基于 token 的模型,这就像让一个只懂莎士比亚的学者去读一份蛋白质晶体衍射图,完全是两个世界。
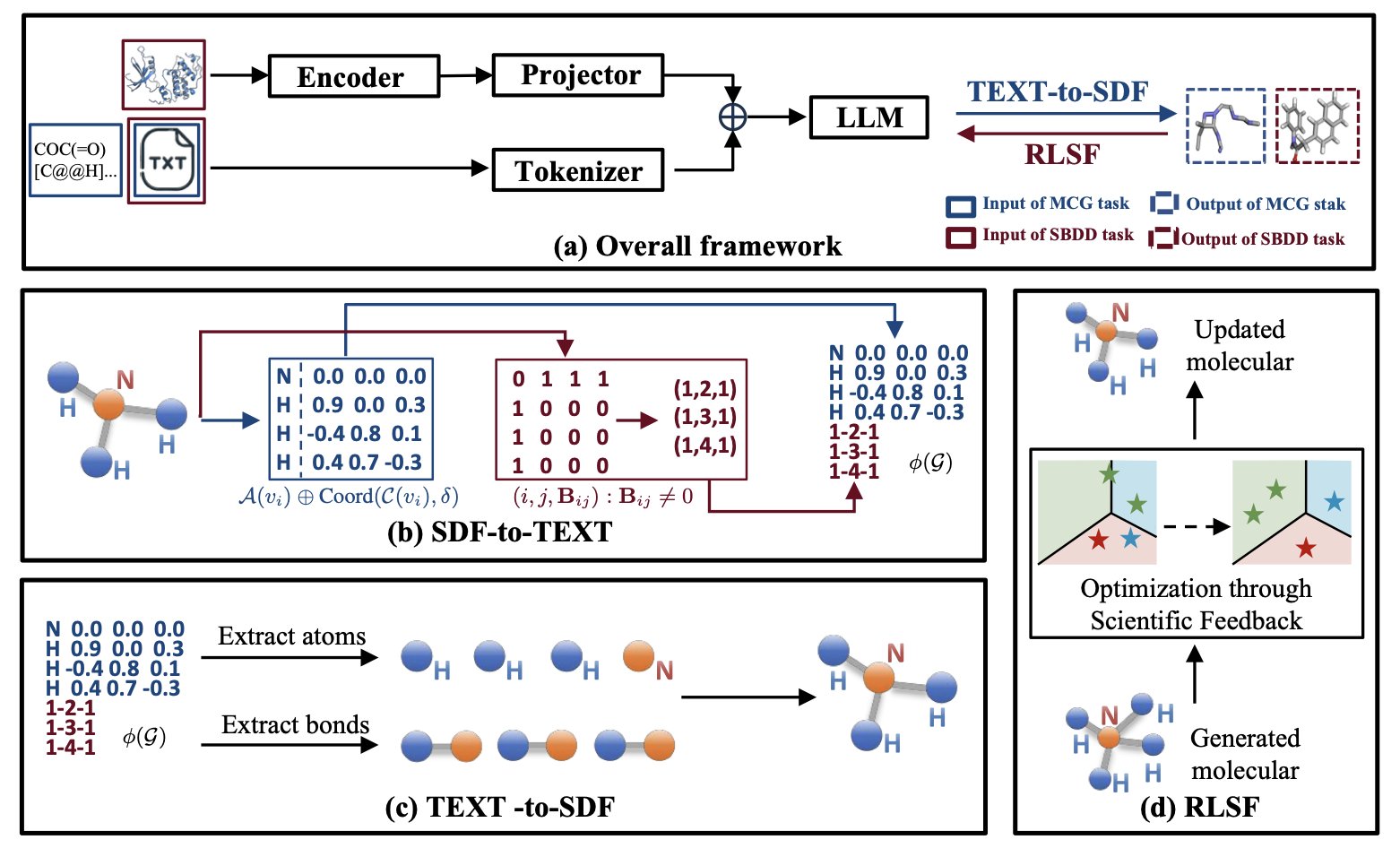
Chem3DLLM 的核心,研究者们称之为「可逆压缩分子标记化」(RCMT)。
这听起来有点拗口,但可以把它想象成一个翻译器。它能把一个复杂的 3D 分子雕塑,精准地转换成一本薄薄的、纯文本的「组装说明书」。LLM 看不懂雕塑,但它能读懂说明书。
最关键的是,「可逆」意味着这个过程是无损的——你可以根据这本说明书,百分之百地还原出原来的那个 3D 雕塑,不多一个原子,也不少一根键。这个技术把分子数据的大小压缩了 3 倍,对于动辄处理海量数据的我们来说,这本身就是个不小的工程进步。
解决了输入问题,接下来就是怎么让模型干活了。
在基于结构的药物设计(SBDD)里,我们关心的是两件事:蛋白口袋和配体分子。
过去的方法通常是各管一摊,或者用复杂的图模型来处理。Chem3DLLM 厉害的地方在于,它把这两件事放在一个统一的 LLM 架构里解决了。它用一个轻量级的投影模块,把 3D 的蛋白口袋特征也「翻译」并对齐到 LLM 能理解的语义空间里。这样一来,模型就能同时「看到」口袋长什么样,以及它自己生成的分子长什么样,然后在一个统一的框架内进行端到端的学习。
当然,只让 LLM 自由发挥,它可能会给你生成一些化学上根本不存在的「怪物」。这就引出了另一个亮点:基于科学反馈的强化学习(RLSF)。这部分就像给模型请了个严格的化学和物理老师。模型每生成一个构象,这个「老师」就会根据化学键的稳定性、能量高低这些基本物理化学原理给它打分。生成了稳定的、低能量的分子,就给奖励;生成了奇形怪状、能量极高的东西,就给惩罚。通过这种方式,模型学会的就不只是模仿数据,而是遵循真实的物理化学规律去创造。
结果怎么样?
📜Paper: https://arxiv.org/abs/2508.10696v1
3. AuroBind:从虚拟筛选到皮摩尔级命中
这些年,各种「突破性」的虚拟筛选(VS)工具声称它们的 AI 能以前所未有的精度预测分子对接。它们确实很擅长生成漂亮的图片——一个小分子完美地嵌在蛋白的结合口袋里,每一个氢键都恰到好处。问题是,这些图片里有一半的分子,在实验里的结合亲和力可能还不如一根湿面条。
这就是我们这个领域最痛苦的现实:一个完美的对接构象(pose),和一个有活性的分子之间,隔着一条鸿沟。
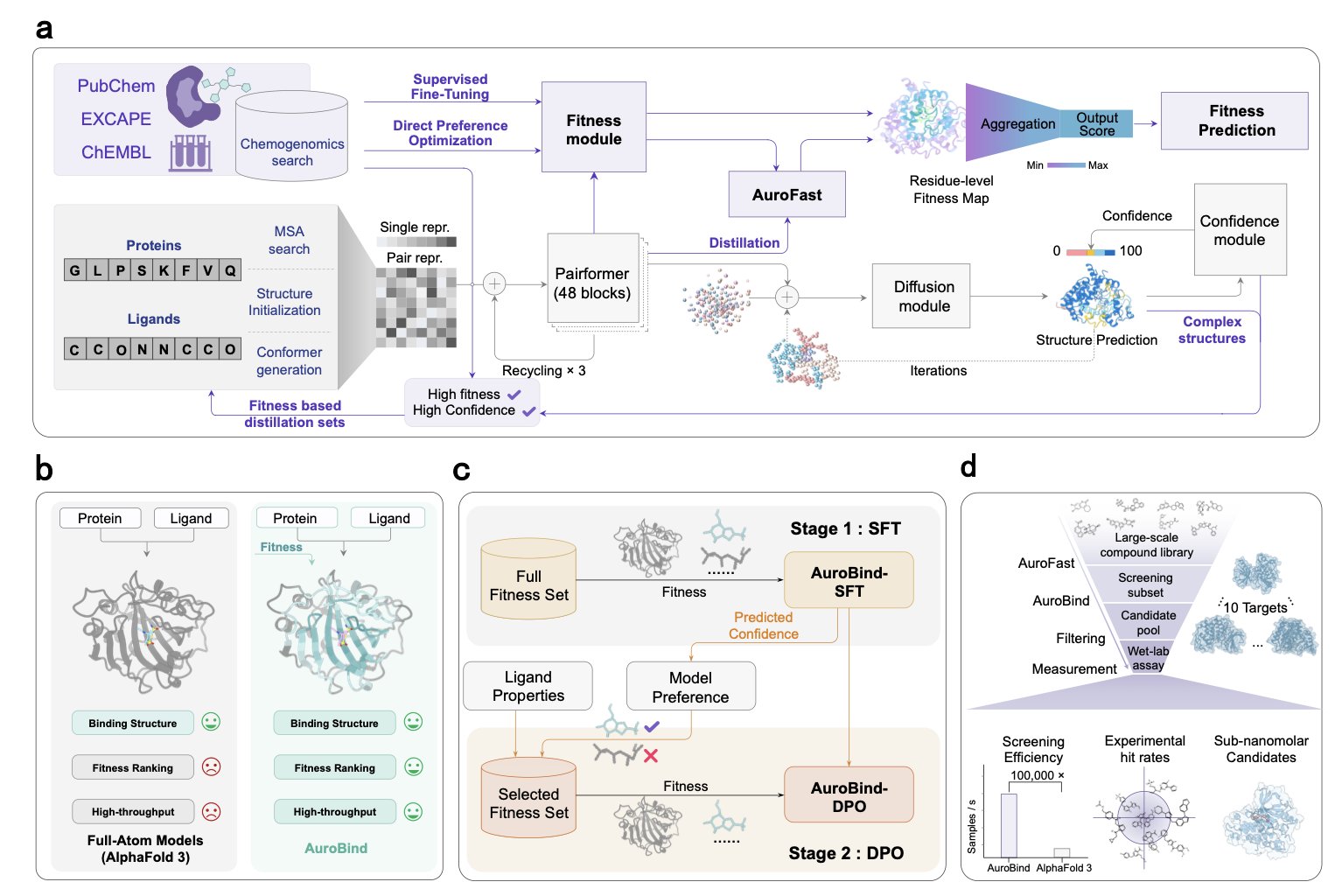
AuroBind 的作者们显然对这种痛苦有切身体会。他们没有再搞一个只会「看图说话」的模型,而是从根子上解决问题。
他们的做法分两步:
1. 学规矩 :首先,他们用海量的、高质量的蛋白 - 配体复合物晶体结构数据来训练模型。这一步是基础,是教会 AI 正确的「几何规则」,让它知道一个分子应该以什么样的构象待在口袋里才算合理。
2. 学品味 :这是真正的点睛之笔。他们在第一步的基础上,用一个包含了数百万个化合物 - 靶点活性数据的巨大数据库,对模型进行「微调」。这一步不再是教 AI「怎么放」,而是教它「放什么」。模型被迫去学习,什么样的结构特征和相互作用,才真正对应着强大的生物活性。它学会了区分「看起来不错」和「真的很好」。
结果在十个完全不同的蛋白质靶点上,实验验证的命中率高达 7% 到 69%。这在虚拟筛选领域,是一个非常、非常惊人的数字。而且,找到的化合物里不乏亚纳摩尔甚至皮摩尔级别的猛药。
但这还不是最厉害的。
这篇论文里真正的「炸裂时刻」,是他们在「孤儿」GPCR 靶点上的前瞻性筛选。比如 GPR151,这种靶点没有任何已知的晶体结构,甚至连一个已知的结合物都没有。对于传统的 VS 方法来说,这简直就是在一片漆黑的、没有地图的森林里找一根特定的绣花针。而 AuroBind 就在这种极限条件下,成功地找出并验证了全新的、有活性的分子。
这证明了它不只是一个更擅长「内插」的工具,它具备了真正的「外推」能力——去探索我们从未涉足过的化学空间。
最后,为了让这一切变得实用,他们还搞出了一个叫 AuroFast 的「学生模型」。通过知识蒸馏,这个轻量级模型在保持精度的同时,把筛选速度提高了十万倍。这意味着,过去需要一个计算集群跑几个月的任务,现在可能几个小时就搞定了。
📜Paper: https://arxiv.org/abs/2508.02137