
目录
- 最新的 AI 模型在从零开始生成三维分子方面取得了长足进步,但它们仍然像一个热情有余、经验不足的建筑师,造出的分子在图纸上看似合理,却常常违背基本的物理和化学定律,需要大量「后期装修」才能勉强入住。
- 面对药物筛选数据中海量的「无效」化合物,通过人为将活性与非活性化合物的比例调整至 1:10,可以显著提升 AI 模型的预测能力,使其从噪音中发现真正的信号。
- AI 在药物发现领域的应用正在告别「一招鲜」的炒作,演变为一个包含多种专用工具的成熟工具箱,精准解决从靶点识别、分子对接、成药性预测到化学合成等一系列实际问题。
- CROP 通过一个「前缀」机制,让大型语言模型在「阅读」分子序列的同时,也能「看见」其二维拓扑图和三维空间构象,从而极大地提升了它们对化学世界的理解力。
- AlphaFold 3 是一个惊人的结构预测工具,但这项研究清晰地表明,它是一个需要被精确告知生物学背景的精密仪器,而非一个能读懂你心思的魔法盒子。
1. AI 分子设计:看上去很美,物理学不答应
能不能让一个 AI 程序,像一个灵感无限的化学家一样,从零开始,凭空「想象」出全新的、具有成药潜力的小分子?
这就是所谓的「无条件生成」(unconditional generation)。你不需要给它一个靶点,不需要给它任何约束,你只要对它说:「去吧,给我创造一个像药的分子」,然后就等着看奇迹发生。
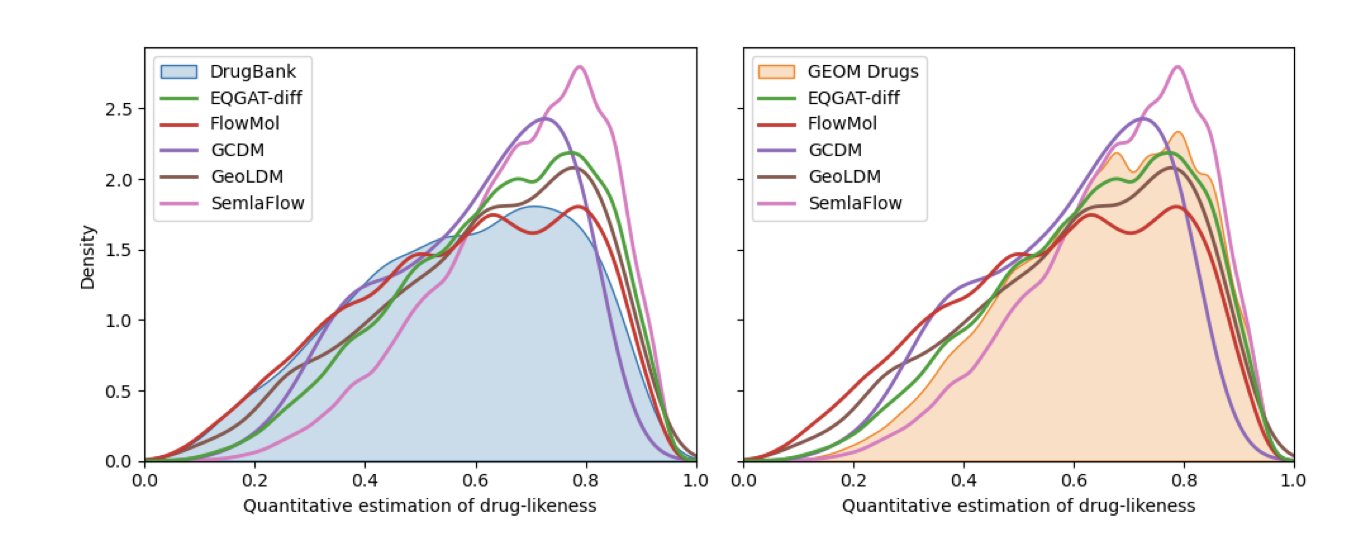
这篇在 ICLR 研讨会上发表的论文,就对当前五个最先进的「造梦机器」(EQGAT-diff, FlowMol, GCDM, GeoLDM, 和 SemlaFlow)进行了一次严格的检验。
好消息是,我们确实进步了。
几年前,这类程序吐出来的东西,大部分都是一堆原子和化学键的胡乱拼接,让任何一个有机化学家看了都想把电脑砸了。而现在,最好的选手 SemlaFlow,已经能做到在没有任何人工干预的情况下,它生成的分子里有 87% 是「有效」的。
这里的「有效」,意思基本上是「至少在二维图纸上看起来像个正经分子,原子价态正确,化学键没画错」。
这篇论文最核心的贡献,是建立了一套非常严苛的「质检流程」。它们不仅检查分子在二维图纸上是否合理,还动用了像 RDKit 和 PoseBusters 这样的工具,去检查三维结构中的物理现实:键长是不是太长或太短?键角是不是扭曲到了一个不可能的角度?有没有两个原子被硬生生地塞在了一起,产生了立体冲突(steric clash)?
这就像是审查一个建筑师的图纸。你不仅要看平面图上房间和门窗的位置对不对(二维有效性),你还要去检查三维模型,看看楼梯的坡度是不是陡到能摔死人,承重墙是不是薄得像纸一样(三维物理合理性)。
经过这套严苛的质检,AI 们的「初稿」就暴露出了很多问题。这也解释了为什么「后处理」如此重要。所谓的后处理,其实就是让一个懂行的「老法师」(比如用 RDKit 进行能量最小化),去把 AI 画出来的那些异想天开的结构,硬是掰回到一个物理上比较合理的状态。经过这么一番「装修」,几乎所有模型的「合格率」都提升了。比如 GCDM,它的合格率能从不怎么样的水平,一跃提升到 95.2%。但这恰恰反过来说明,它一开始交出的毛坯房有多粗糙。
另一个有趣的发现是,这些 AI 虽然学到了很多关于「药物应该长什么样」的统计学知识(比如 Lipinski 五规则),但它们似乎还有点「胆小」。它们生成的分子,大都和训练集(GEOM Drugs 数据集)里的分子长得很像,不敢大胆地去探索更广阔、更新颖的化学空间。它们像是一个只会模仿大师风格的画匠,而不是一个能开创自己画派的艺术家。
无条件 3D 分子生成的能力确实在飞速发展,但我们离那个「一键生成完美候选药物」的按钮还很远。AI 目前还只是一个强大的灵感生成器,它能给你很多看似不错的想法。但要把这些想法变成现实,你仍然需要一个经验丰富的化学家,带着对物理定律的敬畏,去审视、修正、并最终决定,哪些想法值得我们花上数月的时间和精力,在实验室里把它真正地做出来。
📜Title: An Evaluation of Unconditional 3D Molecular Generation Methods
📜Paper: https://openreview.net/forum?id=ZU1m9Y4tYy
2. AIDD:1:10 的数据配比才是真金
我们对高通量筛选(HTS)数据是又爱又恨。爱的是,我们终于有了海量的数据点,看起来像是进入了「大数据」时代。恨的是,这些数据绝大多数都是垃圾。一场几十万个化合物的筛选下来,能有几个活性不错的苗头就算烧高香了。这就像在一条全是泥沙的河里淘金,你筛出来的绝大部分都是泥(非活性化合物),真正的金子(活性化合物)少得可怜。
现在,每个人都在谈论用 AI 来加速这个淘金过程。但问题是,如果你直接把这满是泥沙的原始数据喂给一个机器学习模型,会发生什么?
模型会学到一个非常简单但完全没用的策略:不管看到什么,都预测它是「泥沙」。这样做,它的准确率可以轻松达到 99.9%,因为它猜对了绝大多数情况。但对于想找金子的人来说,这个模型一文不值。
我们该如何调整训练数据中「金子」和「泥沙」的比例。
他们使用了一种叫做「K-ratio 随机欠采样」的方法。
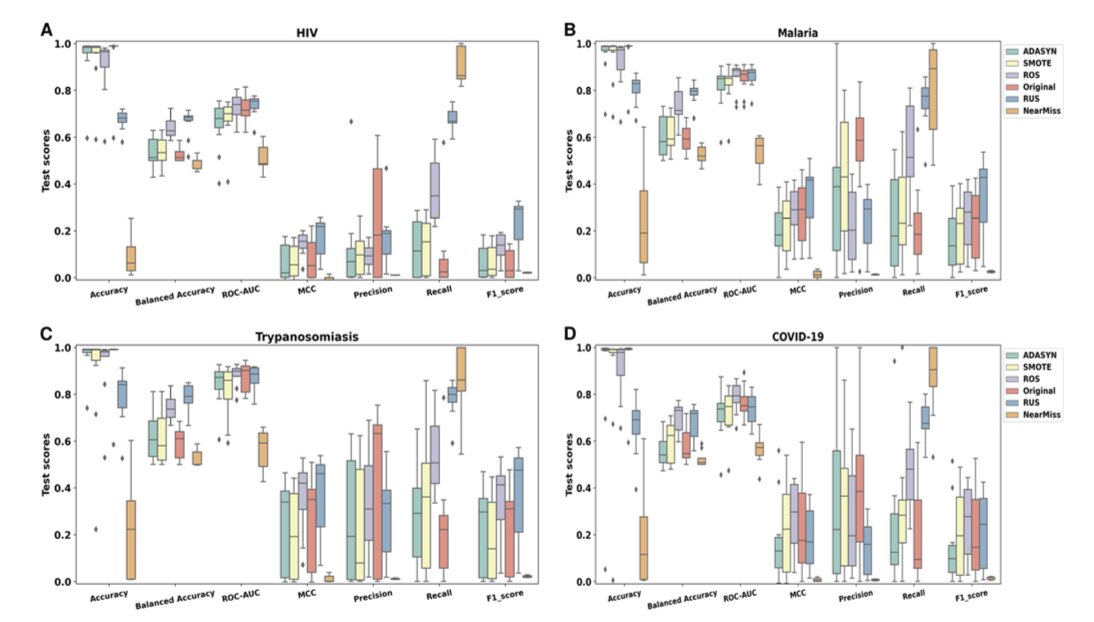
简单来说,就是固定住所有宝贵的活性化合物,然后从海量的非活性化合物中,按不同的比例随机抽取一部分来组成训练集。他们测试了从 1:1(完全平衡)到 1:1000(接近原始数据)的各种比例。
结果当比例设定在 1:10 时,也就是一个活性化合物配十个非活性化合物,几乎所有模型的性能都达到了峰值。
如果非活性化合物太多(比如 1:1000),模型就被「淹没」了,学不会识别活性分子的精细特征。如果非活性化合物太少(比如 1:1),模型又会损失大量关于「什么不是我们想要的」的宝贵负面信息,导致它可能会把很多看起来有点像金子的黄铜(假阳性)也当成宝贝。1:10 的比例,恰好给了模型足够的负样本来学习边界,又不会让它被噪音带偏。
研究者还深入分析了模型犯错的原因。他们发现,当一个活性分子和一个非活性分子的化学结构长得特别像时(我们称之为「活性悬崖」,activity cliff),模型就很容易被搞糊涂。这不是算法的错,这恰恰是药物化学的现实。一个微小的结构改动,就可能让一个药物从「灵丹」变成「废铁」。
数据平衡不是万能药。你的化合物库本身的多样性至关重要。如果你的库里塞满了成千上万个长得差不多的类似物,那你只是在给 AI 模型出难题。
📜Title: Adjusted Imbalance Ratio Leads to Effective AI-Based Drug Discovery Against Infectious Disease
📜Paper: https://www.nature.com/articles/s41598-025-15265-5
3. AI 药物设计:从黑箱到工具箱的进化

目前我们对人工智能(AI)的情感可以说是相当复杂。一方面,我们看到了它颠覆性的潜力;另一方面,我们也见证了太多华而不实的炒作。每隔几年,就会有一种新的计算方法号称要让药物化学家集体失业,但第二天我们还是得老老实实地回到通风橱前,处理那些粘稠的、不听话的化学反应。
Journal of Cheminformatics 最近推出的这期「AI 药物发现」特刊,给人的感觉不再是那种「AI 将统治一切」的宏大叙事,而更像是一群经验丰富的工程师,在向你展示他们为解决特定问题而精心打磨的各式工具。
AI 正在从一个神秘的「黑箱」,变成一个我们看得懂、用得上的「工具箱」。
第一格抽屉:重新定义「看靶点」
传统上,我们做基于结构的药物设计(SBDD),前提是你得有一个蛋白质的三维结构,最好是带配体的晶体结构。没有它,大部分计算工具就成了睁眼瞎。但这期特刊里提到的 CLAPE-SMB 方法,就试图绕开这个限制。它能仅凭蛋白质的氨基酸序列,就预测出 DNA 的结合位点。这好比你不需要亲眼看到一把锁,只要拿到它的设计图纸(序列),就能大致推断出钥匙孔(结合位点)在哪里。这对那些难以结晶的靶点来说,无疑打开了一扇新的大门。
而在我们更熟悉的分子对接领域,工具也在升级。我们都知道,传统的对接软件打分函数(scoring function)有多不靠谱。Gnina v1.3 把卷积神经网络(CNN)用到了这个环节。这就像是从用一把尺子生硬地测量配体和口袋的匹配度,升级到了请一位经验丰富的木工(CNN)来凭手感和经验判断这个榫卯结构是否严丝合缝。它甚至还开发了共价对接的打分功能,这在以前可是个老大难问题。
更有意思的是像 PoLiGenX 这样的工具,它体现了一种新的合作哲学。它允许你先给 AI 一个「参考分子」,告诉它:「我知道长成这样的分子能很好地嵌在这个口袋里,你能不能在这个基础上,帮我变出一些更好的新分子?」这承认了计算机并非全知全能,人类化学家的直觉和经验依然宝贵。AI 不再是高高在上的「设计者」,而更像一个能举一反三、不知疲倦的「高级助手」。
第二格抽屉:从「算命」到「天气预报」
ADMET(吸收、分布、代谢、排泄、毒性)预测是 AI 最早大显身手的领域之一,但一直有个问题:模型只告诉你一个结果,却不告诉你它有多自信。
一个模型说某个分子「无毒」,另一个模型说它「有毒」,你该信谁?
这期特刊里的 AttenhERG 和 CardioGenAI 等新模型,正在解决这个问题。它们不仅预测 hERG 毒性或心脏毒性,更重要的是,它们开始引入不确定性评估。这就像天气预报从简单的「明天晴天」,进化到了「明天晴天,降水概率 10%」。后者显然更有用,因为它让你知道风险有多大。通过使用贝叶斯方法等策略,这些模型会告诉你:「我预测这个分子无毒,并且我对这个预测有 95% 的把握。」这种带有「自信度」的预测,对于我们决定是否要在一个项目上投入数百万美元来说,价值千金。
第三格抽屉:AI 终于开始关心「怎么做菜」了
这是过去所有 AI 设计软件最大的短板。一个 AI 可以给你设计出一个理论上能治愈癌症、结构上美如天仙的分子,但当你把这个结构拿给合成化学家时,他可能会直接把图纸扔回你脸上,因为它儿在地球上根本做不出来。
现在,AI 终于开始正视这个「合成可行性」的问题了。特刊里讨论了新的反应预测基准和策略。这还不够,一些研究开始利用大型语言模型(LLMs)去「阅读」海量的化学专利和文献。这就像是让 AI 去学习人类历史上所有的「菜谱」,不仅要学会照着做,还要理解背后的「烹饪原理」,从而为 AI 设计出的新分子,规划出一条现实可行的「烹饪路线」。
AIDD 正在褪去浮华,变得越来越「接地气」。它不再试图成为一个无所不能的「大神」,而是把自己拆解成一系列可以解决真实世界问题的工具。
📜Title: Advanced machine learning for innovative drug discovery
📜Paper: https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01061-w
4. AI 分子模型长了眼睛
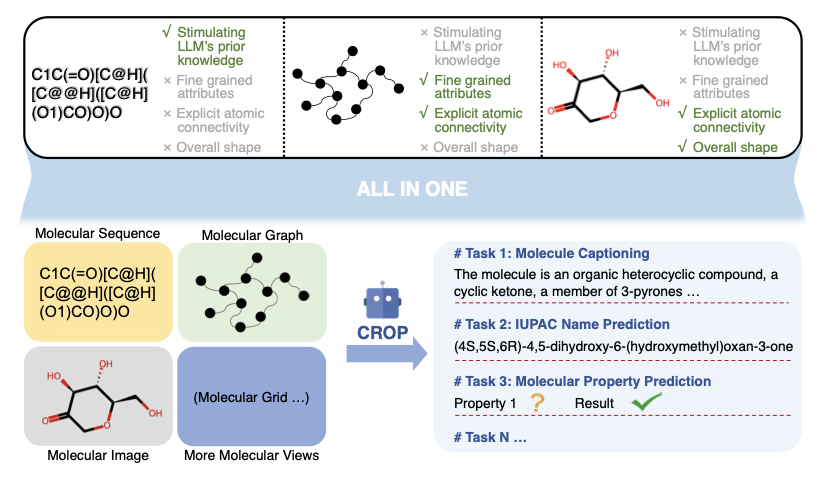
在过去的几年里,大型语言模型(LLMs)席卷了几乎所有领域。在化学界,我们自然也想让这些「语言学家」来帮我们读懂分子的语言。我们最常用的分子语言,就是 SMILES 字符串——一串描述了分子结构的文本。
但这一直有个根本性的问题。SMILES 就像是你在用纯文本,去跟一个盲人描述梵高的《星空》。你可以告诉他:「画面里有很多黄色的小漩涡,背景是深蓝色。」但他永远无法真正「看见」那幅画的构图、笔触和空间感。
同样,一个只读 SMILES 的 LLM,它知道哪些原子和哪些原子相连,但它对这个分子在三维空间里究竟长成什么样,哪个基团朝上,哪个基团朝下,其实是一无所知的。
CROP 架构,就是想给这个「盲人语言学家」安上一双眼睛。
它的想法是让 LLM 阅读那段描述《星空》的文字(SMILES)之前,我们能不能先给它看一眼画的草图(分子的二维拓扑图),再让它摸一下这幅画的立体浮雕版(分子的三维空间构象)?
当然,这里面真正的挑战在于工程实现。
LLM 的「工作记忆」(也就是它的上下文窗口)是有限的。你不能直接把一张高清大图和一张复杂的网络图,硬塞给一个习惯了处理文本的程序。
CROP 设计了一个非常高效的「预处理器」,叫做「SMILES 引导重采样器」。这个东西的作用,就是把那张草图和那块浮雕,intelligently 压缩成一段非常简短的、包含了最关键信息的「内容提要」,我们称之为「前缀」。然后,它把这段「内容提要」放在 SMILES 字符串的前面,一起喂给 LLM。
这样一来,LLM 在开始阅读那段枯燥的文本描述之前,就已经先对这幅画的整体构图和空间感,有了一个大致的印象。这个压缩过程,还是被 SMILES 本身所「引导」的。这意味着,LLM 会利用它自己对文本的初步理解,回头去告诉那个预处理器:「嘿,我觉得这幅画里,那个左上角的大漩涡好像很重要,你在做内容提要的时候,多给它一点笔墨。」这是一个非常优雅的反馈循环。
那么,长了眼睛的 LLM,表现如何呢?
结果是,它几乎在所有任务上都成了一个更好的化学家。
比如在「分子描述」(molecule captioning)这个任务上,也就是让模型看着一个分子,然后用自然语言描述它的特征。CROP 的表现远超其他模型。这说明,当它同时看到了分子的连接方式和空间形状后,它能更深刻地理解这个分子的「性格」,并用更准确的语言把它描述出来。
作者们还做了「消融实验」来验证他们的想法。他们试着只给模型看草图,或者只给它摸浮雕。结果发现,效果都不如两者都给的时候好。这证明了,二维拓扑和三维空间,就像是左眼和右眼,它们提供的是互补的、缺一不可的信息,只有两者结合,才能形成一个完整、立体的认知。
📜Title: CROP: Integrating Topological and Spatial Structures via Cross-View Prefixes for Molecular LLMs
📜Paper: https://arxiv.org/abs/2508.06917v1
5. AlphaFold 3 的现实检验:它不是魔法
自从 AlphaFold 问世以来,结构生物学和药物发现领域的人们,仿佛突然从石器时代跨越到了太空时代。那些我们曾需要耗费多年时间、巨额经费和博士生心力才能解析的蛋白质结构,如今似乎只需将序列输入计算机,悠闲地喝杯咖啡,就能获得一个「scarily good」的模型。AlphaFold 3 的推出,更是将这种能力扩展至蛋白质与其他分子之间的相互作用上。
但总有一个挥之不去的问题:这些模型预测的,都是教科书上那种最标准、最纯净的蛋白质。可现实世界是 messy 的。同一个蛋白质,在不同菌株、不同物种、甚至不同个体之间,总会有一些细微的氨基酸差异。我们称之为「天然变体」。
那么,AlphaFold 3 能处理好这种真实世界的复杂性吗?它能分辨出一个氨基酸的改变,是如何微妙地影响蛋白质局部构象的吗?
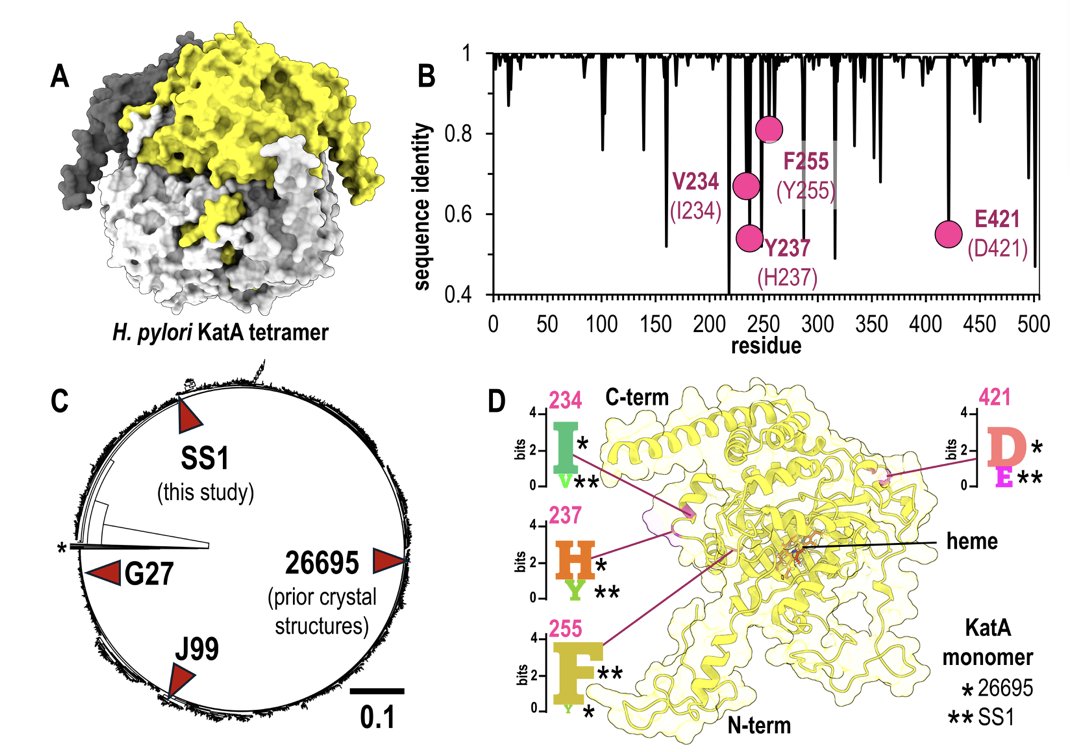
这篇研究试图来回答这个问题。他们选择了一个研究得很透彻的「老朋友」——来自幽门螺杆菌的过氧化氢酶 KatA。我们已经有了来自 26695 菌株的 KatA 的高清晶体结构。于是,研究者们又费心去解析了另一个菌株(SS1)的 KatA 晶体结构,这个新版本与老版本之间有几个氨基酸的差异。
这就构成了一个完美的实验设置。他们手里有了一个全新的、AlphaFold 3 的训练集里没有的「考题」,更关键的是,他们还有一份由实验得出的、百分之百正确的「标准答案」。
接下来,他们就开始「考试」了。
当他们把 SS1 菌株的序列,连同正确的生物学信息——「嘿,AlphaFold,它儿在细胞里是个四聚体」——一起输入模型时,结果让人印象深刻。预测出的结构和他们辛辛苦苦解析的晶体结构,在总体上和大部分细节上都吻合得非常好。这再次证明了 AlphaFold 3 的强大。
但好戏在后头。
他们接着做了一个小小的「破坏性」测试。他们故意「欺骗」AlphaFold 3,告诉它 KatA 是一个单体,而不是四聚体。这就像是给了建筑师一套四合院的图纸,却告诉他你只想盖一间茅草屋。
结果模型预测出的总体折叠看起来还像那么回事,但当你放大去看那些发生突变的、以及本应形成四聚体界面的关键区域时,一切都乱套了。氨基酸侧链的朝向,差之毫厘,谬以千里。
这才是这项研究最核心的贡献。它不是在说 AlphaFold 3 不好,恰恰相反,它是在教我们如何正确地使用这个强大的工具。它用最直接的证据告诉我们:AI 并没有让生物学家变得多余,反而让深刻的生物学知识变得前所未有的重要。你必须先知道你的蛋白质在细胞里到底是怎么回事——它是单打独斗,还是和三个兄弟抱团取暖?——你才能向 AI 提出一个有意义的问题。
这项工作也像是在为传统的实验结构生物学正名。如果没有研究者们解析的那个新的 SS1 晶体结构作为「地面真实」,整篇论文的讨论都将是空中楼阁。我们仍然需要有人在实验室里,日复一日地进行那些看似枯燥的纯化、结晶和 X 射线衍射工作。因为正是这些实验数据,构成了训练和验证 AI 模型的基石。
📜Title: AlphaFold 3 accurately models natural variants of Helicobacter pylori catalase KatA
📜Paper: https://journals.asm.org/doi/10.1128/spectrum.00670-25