
目录
- 通过从头计算设计,研究者成功将难搞的膜蛋白 CCR8 改造为水溶性蛋白,同时保留了关键的抗体结合位点,为 GPCR 药物研发扫清了一大障碍。
- SEAL 通过将分子拆解为化学基团并独立分析其贡献,让图神经网络的预测结果变得直观可信,解决了 AI 模型的「黑箱」问题。
- 通过即插即用的结构和属性控制模块,CMCM-DLM 让 AI 分子生成模型学会了「戴着镣铐跳舞」,在不重新训练的条件下实现精准的定制化分子设计。
1. 计算设计可溶性 GPCR,攻克 CCR8 成药难题
GPCR(G 蛋白偶联受体)家族就像一座金矿,超过三分之一的已上市药物都靶向它们。但同时,它也是出了名的难开发。因为它是膜蛋白。
你可以把膜蛋白想象成一块镶嵌在肥皂泡(细胞膜)上的油腻的石头。它天生就喜欢待在油性环境里。一旦你试图把它从细胞膜里拽出来,放到水溶液里,它就会变得极不稳定,像受惊的猫一样蜷缩成一团,失去原有的结构和功能。这就给我们后续的工作,比如筛选抗体、做生化实验、解析结构,带来了无穷无尽的麻烦。
这篇论文要处理的就是这个问题,他们选了一个当下非常热门的免疫肿瘤学靶点——CCR8。CCR8 主要在肿瘤浸润的调节性 T 细胞(Tregs)上高表达,是清除肿瘤微环境中「叛徒」T 细胞的理想靶点,各大药厂都在布局。
怎么把「疏水」变成「亲水」?
研究者们没有用传统的试错法去修修补补,而是直接用计算设计的方法,釜底抽薪,给 CCR8 做了一次「脱胎换骨」的大手术。
他们的思路是这样的:
1. 保留核心功能区 :首先,他们明确了目标。他们不指望这个改造后的蛋白还能传递信号,他们只需要它能被特异性的抗体(mAb1)识别就行。这意味着,只要保留住抗体结合的那个关键表面区域(表位)的正确构象就可以了。
2. 全面替换其他部分 :对于蛋白的其他部分,特别是那些原来深埋在细胞膜里的「油腻」区域,他们用上了像 ProteinMPNN 这样的 AI 模型。这个模型干的活儿,就是把那些疏水的氨基酸,大规模地替换成亲水的氨基酸。
3. 确保结构稳定 :当然,不能瞎换。整个替换过程需要 AlphaFold 这样的结构预测模型全程「监工」,确保新的氨基酸序列还能折叠成我们想要的那个三维结构,而不是一堆废品。
这就好比,我们要复制一把能开特定锁的钥匙。我们不需要用和原版完全一样的金属材料,我们只需要保证钥匙上「齿」的形状和排列是完全正确的就行了。其他部分,我们可以用更稳定、更容易加工的材料来替代。
结果怎么样?
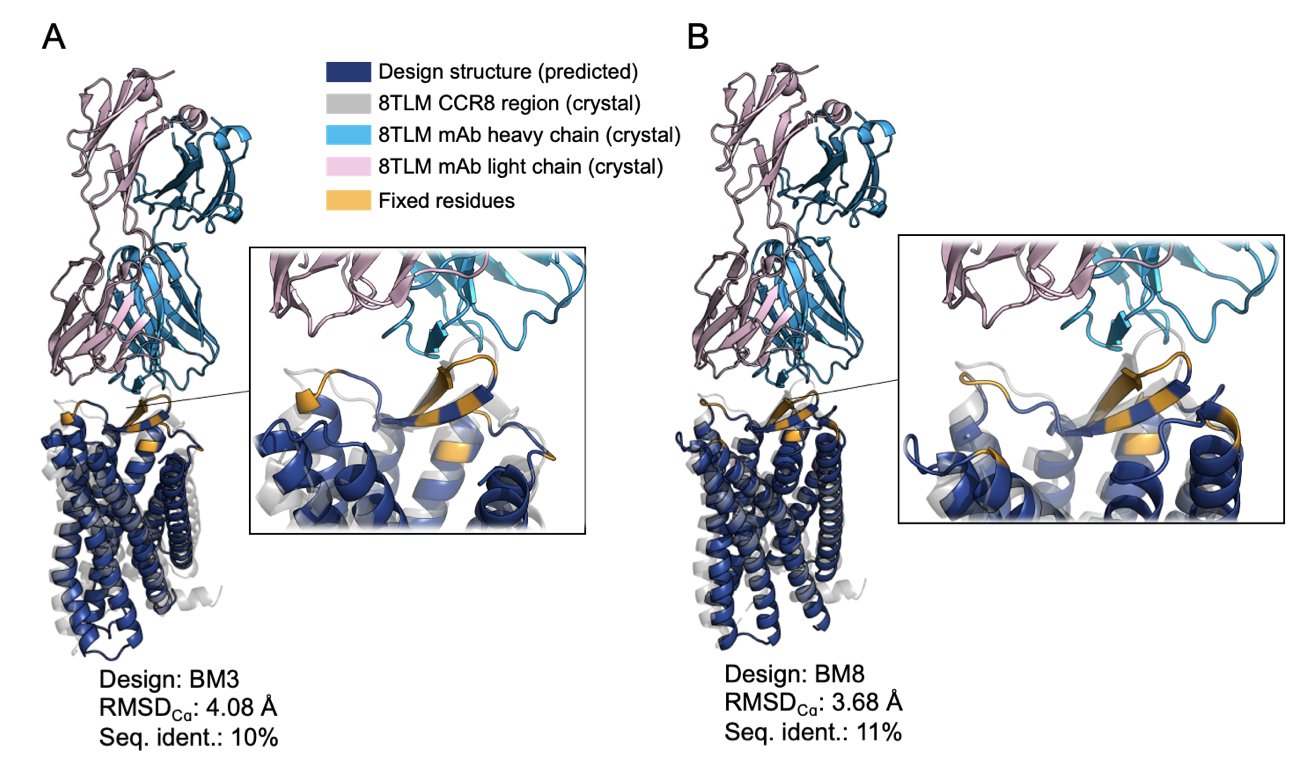
结果他们设计并成功表达了 13 个 CCR8 的水溶性类似物。这些新蛋白的氨基酸序列和天然的 CCR8 相比,相似度只有 10-13%!这基本上等于是一个全新的蛋白质了。
但就是这样一个「面目全非」的蛋白,通过表面等离子共振(SPR)技术检测,它与目标抗体 mAb1 的结合能力(KD 值在 77-857 nM 之间)和野生型 CCR8(KD 值约 190 nM)基本在同一个水平。这证明「钥匙的齿」被完美地保留了下来。
更重要的是,这些蛋白的表达产量能达到 70 mg/L以上。这是一个非常实在的数字,意味着我们可以轻松地获得足够量的、性质稳定的蛋白,用来做各种下游应用。
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.18.670068v1
2. SEAL 模型:让 GNN 的分子预测不再是黑箱
现在谁的项目里没几个 AI 模型在跑。其中,图神经网络(GNN)因为能直接处理分子图结构,成了预测分子性质的香饽饽。
但我们都遇到过同一个尴尬的场景:
项目会上,计算化学家展示了一个模型预测活性超强的分子。作为药物化学家,肯定会问:「为什么?是分子哪个部分让它这么厉害?我下一步该优化哪里?」这时,计算化学家往往只能耸耸肩,说:「模型就是这么说的。」
这就是 GNN 最大的问题——它是个黑箱。
它能给你一个结果,但通常给不了令人信服的理由。这对于需要做出「下一步合成哪个分子」这种关键决策的我们来说,帮助有限。
这篇论文介绍的 SEAL 模型,就是为了模型可解释性而来。
SEAL 是怎么做的?
过去的可解释性方法,很多是在模型预测完之后,再用一些事后归因的方法去「猜」是哪个原子或化学键最重要。这种方法有时会给出一些反直觉的结果。
SEAL 的做法从模型的设计之初就奔着「可解释性」去的。
它的工作原理可以这么理解:一个常规的 GNN 在处理分子时,信息会在整个分子图里传来传去,像一锅粥。一个原子的信息,经过几轮传递,可能会影响到离它很远的另一个原子。这导致很难分清谁是谁的功劳。
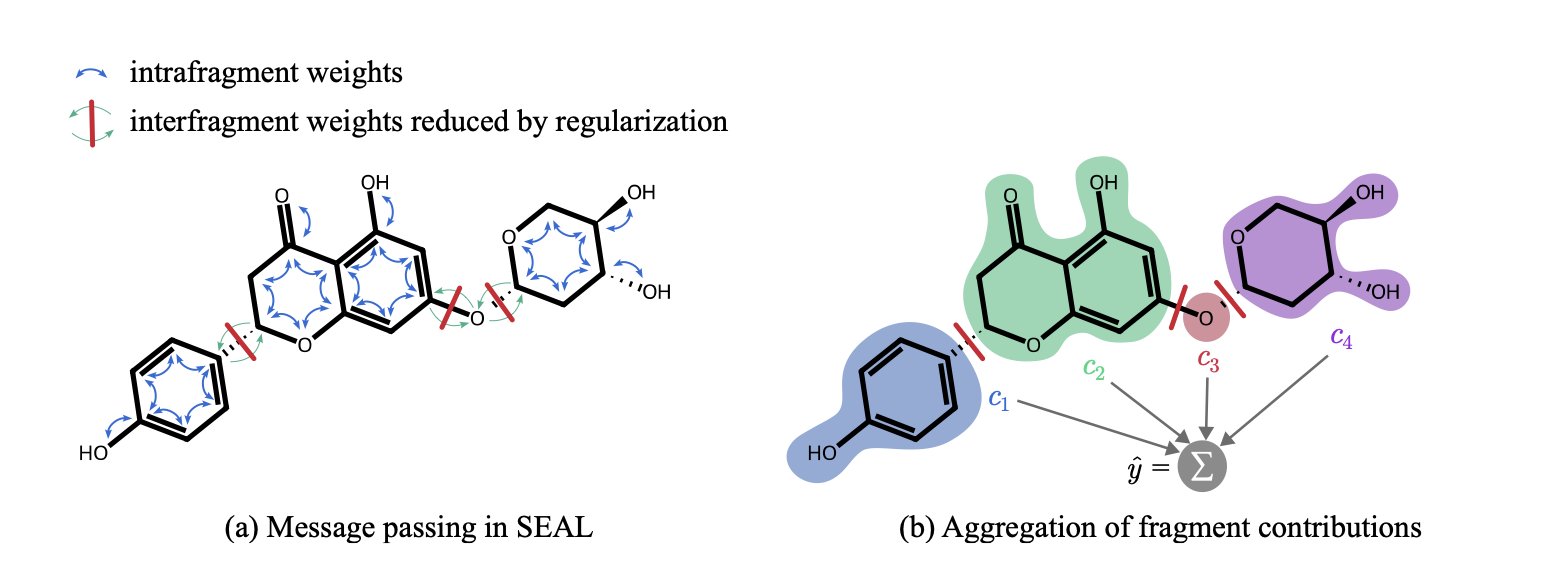
SEAL 则像一个城市规划师。它首先把一个大分子,按照化学家熟悉的官能团、环系等规则,切分成几个有意义的「街区」(化学基团)。然后,它在模型里给这些「街区」之间建起了「防火墙」。它的 GNN 层被特殊设计过,会优先让信息在「街区」内部流通,严格限制信息跨区传播。
这样一来,模型在做预测时,实际上是先独立地评估每一个「街区」(化学基团)对最终性质的贡献,然后再把这些贡献值加起来得到总分。
这在实践中意味着什么?
当 SEAL 告诉你一个分子的溶解度不好时,它还能附上一份「责任清单」:苯环,贡献 -10 分;羧基,贡献 +20 分;这个新接上去的哌啶环,贡献 -30 分。
药物化学家的任务就明确了:看来问题就出在这个哌啶环上,下一轮我们把它换掉试试。
他们不只是在数学上证明了方法的优越性。他们还组织了一场由领域专家参与的用户研究。他们把 SEAL 和其他方法给出的解释,匿名地展示给化学家们看,结果化学家们普遍认为 SEAL 给出的解释更符合他们的化学直 - 觉,也更能帮助他们做出决策。
📜Paper: https://arxiv.org/abs/2508.15015v1
💻Code: https://github.com/gmum/SEAL
3. AI 分子设计:结构和属性终于能兼得了
目前药物研发公司现在手里都或多或少有几个 AI 分子生成模型。这些模型像个灵感无限的初级化学家,能给你画出一大堆新奇的分子结构。
但问题也随之而来,有时候,我们让它围绕一个特定的骨架(scaffold)做优化,结果它画出来的分子虽然性质看着不错(比如溶解度、脂水分配系数都很好),但核心骨架早就被它改得面目全非了。另一些时候,我们强制它必须保留骨架,它倒是听话了,但生成的分子的其他性质又变得一塌糊涂,像块「板砖」,根本没法成药。
这种「结构」和「性质」无法兼得的尴尬,是目前 SMILES 语言基础上的扩散模型的一大痛点。这篇论文提出的 CMCM-DLM,就是来解决这个问题的。
怎么做到既要…又要…?
作者把一个复杂任务拆解成了两个更简单的子任务,并且在生成过程的不同阶段分步执行。你可以把它想象成盖房子:
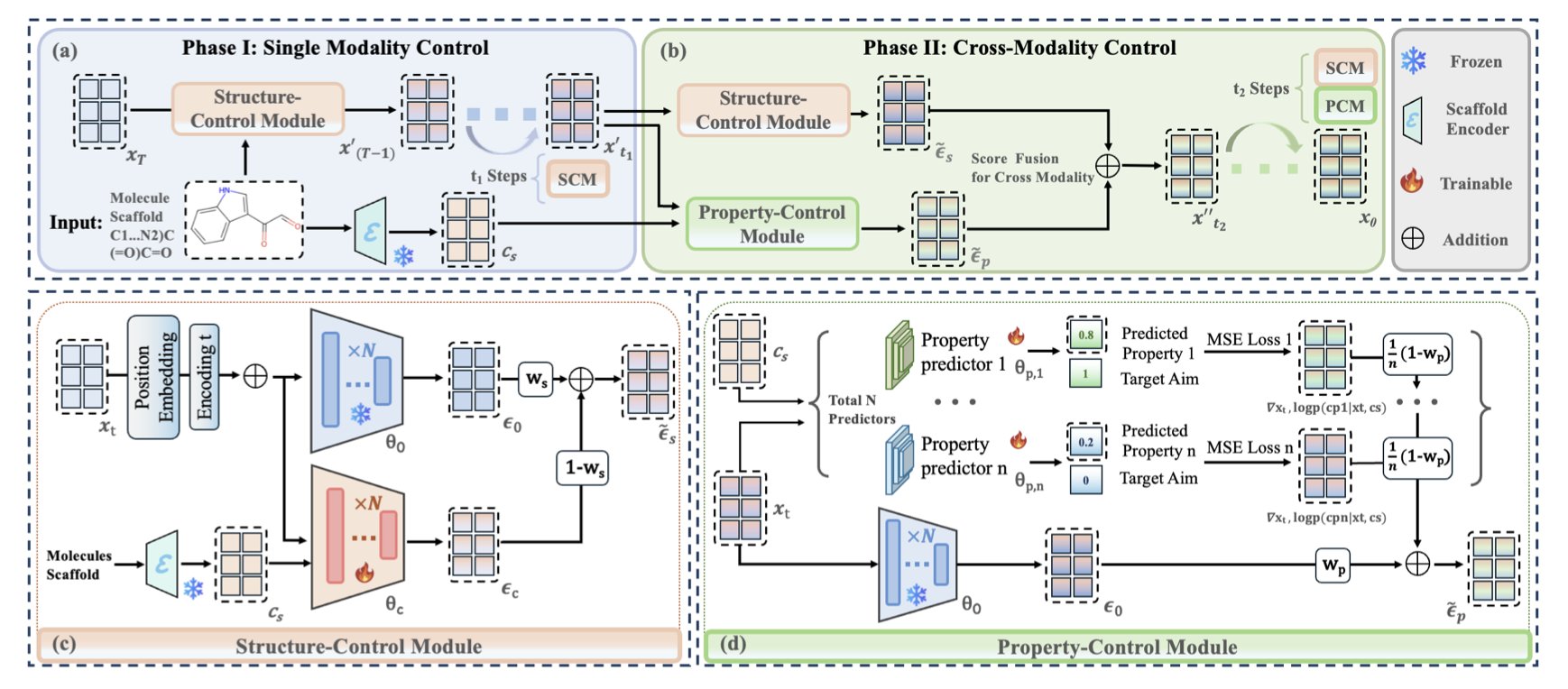
第一阶段:搭框架(结构控制模块 SCM)
在分子生成的早期阶段,整个结构还很模糊,像一团混沌的电子云。这时候,SCM 模块就介入了。它的任务很简单,就是确保这栋房子的承重墙和主体结构(也就是我们想要的分子骨架)必须先立起来,而且位置要对。它在早期就给生成过程上了一个「紧箍咒」,防止结构跑偏。
第二阶段:精装修(性质控制模块 PCM)
当房子的主体框架基本稳定下来后,PCM 模块开始接手。它负责「精装修」,也就是在不破坏承重墙的前提下,去调整细节,比如选择什么样的窗户、铺什么样的地板、用什么颜色的涂料。这些细节,对应的就是分子的各种化学性质,比如 QED(类药性)、PLogP(脂溶性)和 SAS(合成可及性)。PCM 会引导着生成的后期步骤,让最终的分子在满足结构要求的同时,各项性质也达到我们的标准。
即插即用
CMCM-DLM 的这两个控制模块(SCM 和 PCM),被设计成了「即插即用」的插件。这意味着什么?意味着我们不需要为了用这个新功能,而去费时费力地重新训练一个庞大的底层扩散模型。我们可以直接把它俩,像 USB 设备一样,「插」到任何一个已经训练好的模型上。这大大降低了技术转化的门槛,让这个新方法能被快速应用到实际的项目中。
论文里的数据也很有说服力。在多个数据集上,CMCM-DLM 能让生成分子的骨架相似度平均达到 55%,同时目标性质的满足度提升 16%。特别是当需要同时优化多个性质时,它的相对改进能达到 52%。这些都不是小打小闹的提升,对于药物化学家来说,这意味着 AI 给出的建议,废品率更低了,可用的「好点子」更多了。
CMCM-DLM 让我们在做先导化合物优化时,有了一个更听话、也更 AI 助手。它能在我们划定的「规则」(保留核心骨架)内,最大限度地发挥它的创造力,去寻找那个结构和性质都完美的「理想分子」。
📜Paper: https://arxiv.org/abs/2508.14748