
目录
- BioEmu 用生成式 AI 给蛋白质拍「电影」,速度远超传统的分子动力学。它为高通量理解蛋白功能和药物发现,开辟了一条新路。
- 研究者训练了一个模型,让它同时搞定分子重建和属性预测两件事。由此,他们创造出一个「陌生度」指标,既能揪出真正新奇的分子,也提醒我们:什么时候该给 AI 的预测打个问号。
- Mol-R1 用一套迭代训练框架,把大语言模型(LLM)调教成了一个既能给出分子设计答案,又能写出详细化学「解题步骤」的学霸。这让 AI 在药物发现领域的推理能力和可靠性都上了一个台阶。
- PEPBI 数据库第一次给 AI 配齐了「高质量教材」:不仅有蛋白 - 多肽复合物的精细结构图,还附上了精确的实验热力学数据(ΔG, ΔH, ΔS)。AI 终于能学点物理化学了。
- rbiol 让大语言模型拜「虚拟细胞」为师,用模拟出的海量数据,训练出一个懂生物学推理的 AI。它的表现不输、甚至超过了那些用真金白银的实验数据喂出来的模型。
1. BioEmu: AI 让蛋白「动」起来,快过 MD
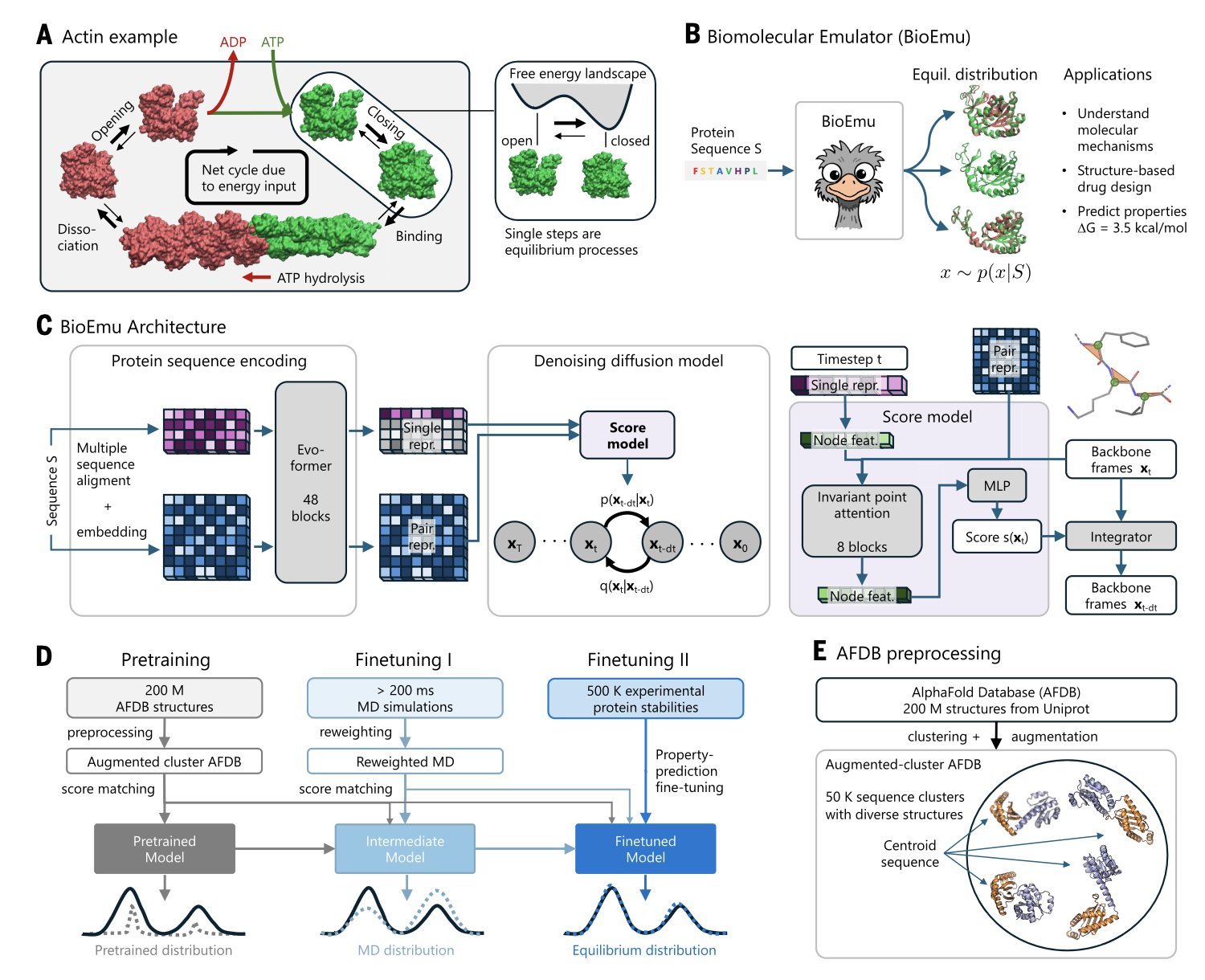
AlphaFold 给了我们一张蛋白质的高清照片,这很了不起。但蛋白质更像一部电影,它会扭动、会呼吸、会改变构象来执行功能。想看这部「电影」,传统方法是分子动力学(Molecular Dynamics, MD),但这就像用一帧一帧手绘的方式做动画,极其耗时,算力成本高得吓人。现在,BioEmu 给我们提供了一台高速摄像机。
研究者把 AlphaFold 当成一个超级阅读器,先用它来「读懂」蛋白质的序列信息,提取特征。然后,他们用一个扩散模型(diffusion model)来「画」出三维结构。这就像一个懂物理规则的艺术家,不是瞎画,而是基于规则生成一整个动态的、处于平衡态的构象集合。为了让模型学得好,研究者们喂给了它海量的数据:超过 200 毫秒的 MD 模拟轨迹和 50 万个实验测定的蛋白质稳定性数据。这相当于让它看完了海量的「教学影片」。
结果它能重现已知的蛋白质构象变化,比如激酶的 DFG-in/out 翻转,在预测蛋白质折叠自由能方面的表现也相当扎实。平均绝对误差小于 0.9 kcal/mol,斯皮尔曼相关系数约0.6。这个精度,足以给评估突变体稳定性的研究者提供靠谱的参考。你可以用它快速筛一遍几百个点突变,看看哪个可能会把你的蛋白搞垮。
BioEmu 能帮我们看到那些在静态结构里「藏」起来的结合口袋。很多变构抑制剂的靶点,就是蛋白质在动态变化中才一闪而过的。BioEmu 让我们有机会批量捕捉这些稍纵即逝的瞬间,为发现新的药物靶点打开了一扇门。
BioEmu 和 MD 是合作关系。它像一个超级「海选」工具,先从无穷的可能性里,快速圈定出一片最有潜力的构象空间。然后,MD 再上场,用精细的、基于物理力场的模拟进行验证,把宝贵的计算资源用在刀刃上。过去只有专业计算团队才能搞定的事,现在实验科学家在自己的工作站上或许就能完成,探索蛋白质动态行为的门槛就这么降下来了。
📜Title: Scalable emulation of protein equilibrium ensembles with generative deep learning
📜Paper: https://www.science.org/doi/10.1126/science.adv9817
2. AI 学会了说「我不知道」
AI 制药领域,一直有个叫「路灯效应」的幽灵在徘徊。
你拿成千上万个已知的激酶抑制剂去训练一个模型,然后让它在海量的虚拟化合物库里淘金。它会给你什么?大概率是一堆跟你投喂的样本长得差不多的「高仿」分子。这就像黑灯瞎火的晚上,你只在路灯底下找钥匙。AI 擅长在光亮处掘地三尺,但真正的宝藏,往往藏在它没见过的黑暗化学空间里。
更头疼的是,AI 自己摸进了「无人区」却毫无知觉。它照样给你一个预测结果,但这个结果的靠谱程度,可能跟掷骰子差不多。我们需要一个工具,让 AI 在踏入未知领域时,能主动举个小旗,告诉我们:「前方路段没信号,我的预测别全信」。
一篇 ChemRxiv 上的预印本论文,就造出了这么一面「小旗子」。
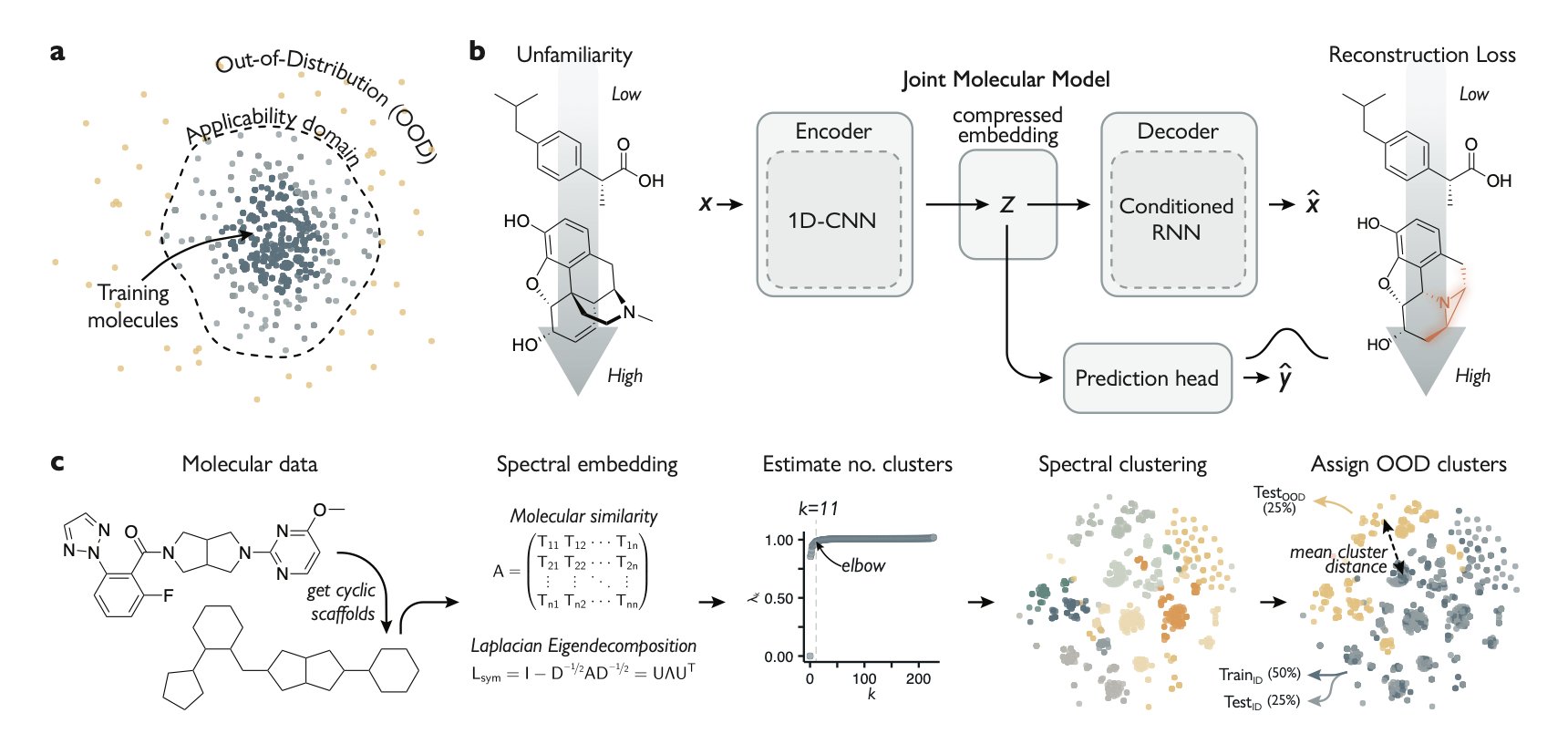
研究者训练了一个「联合分子模型」(Joint Molecular Model)。这个模型得同时学会两件风马牛不相及的事:
1. 预测属性 :判断一个分子有没有活性,这是常规操作。
2. 重建分子 :模型得先把输入的分子压缩成一个包含核心信息的低维「指纹」(这是自编码器的编码过程),再凭着这个「指纹」,把原始分子一模一样地「画」回来(解码过程)。
这好比训练一位文物修复师,既要他能鉴定真伪,还得要求他能把一件打碎的瓷器,凭着碎片复原得分毫不差。
这个模型的精妙之处就在于,当你给它看一个从未见过的、结构清奇的分子时,它在「复原」这一步就会手忙脚乱。因为它在训练中,压根没学过这种「画风」。它复原得有多烂——也就是「重建误差」有多大——就完美地量化了这个新分子对模型而言有多「陌生」(unfamiliarity)。
这面「小旗子」,现在不仅能举起来,上面还带刻度。
这篇论文没有停留在漂亮的计算结果上。研究团队用这个模型,筛选了两个临床上重要的激酶靶点。他们的策略很大胆:专门寻找那些被模型预测为有活性,同时又被标记为高度「陌生」的分子。
这是一次典型的高风险、高回报探索。他们赌赢了。研究者从候选分子中挑了一批,送进湿实验里验证。结果,他们找到了好几个活性达到微摩尔级别的新抑制剂,而这些分子的化学骨架,和训练模型用的那些分子,几乎没有共同点。
他们真的在黑暗里,找到了钥匙。
这项工作,为在广阔化学宇宙中导航的科学家,提供了一个「罗盘」。它补充了现有的「不确定性评估」等工具。现在,我们面对 AI 给出的预测时,不仅可以问「你有多确定?」,还可以问「这东西,你熟不熟?」。
📜Title: Molecular deep learning at the edge of chemical space
📜Paper: https://doi.org/10.26434/chemrxiv-2025-qj4k3-v3
3. Mol-R1: 教会 AI 像化学家一样「思考」
大语言模型(Large Language Model, LLM)像一个天赋异禀但有点叛逆的学生。你甩给他一道复杂的有机化学题,他总能写出正确答案。可你要是问他「怎么想出来的?」,他可能就给你一个眼神,让你自行体会。
这个「黑箱」问题,在新药研发里是要命的。我们需要的不是一个只会报结果的「神谕」,而是一个能一起分析、一起犯错、一起成长的「临床搭子」。
如果 AI 不能解释清楚它为什么觉得某个分子有潜力,我们就没法信任它,更不敢把上百万美元的研发经费,赌在它的一句「我算的」上面。
Mol-R1 就想解决这个问题:让 AI 学会思考,还把思考过程写下来。
没有教科书?我们自己印!
训练 AI 推理,最大的坎是没有一本好的「化学推理教科书」。我们有海量的分子数据库,但很少有数据能把化学家完整的「思维链」(Chain-of-Thought, CoT)记录下来。
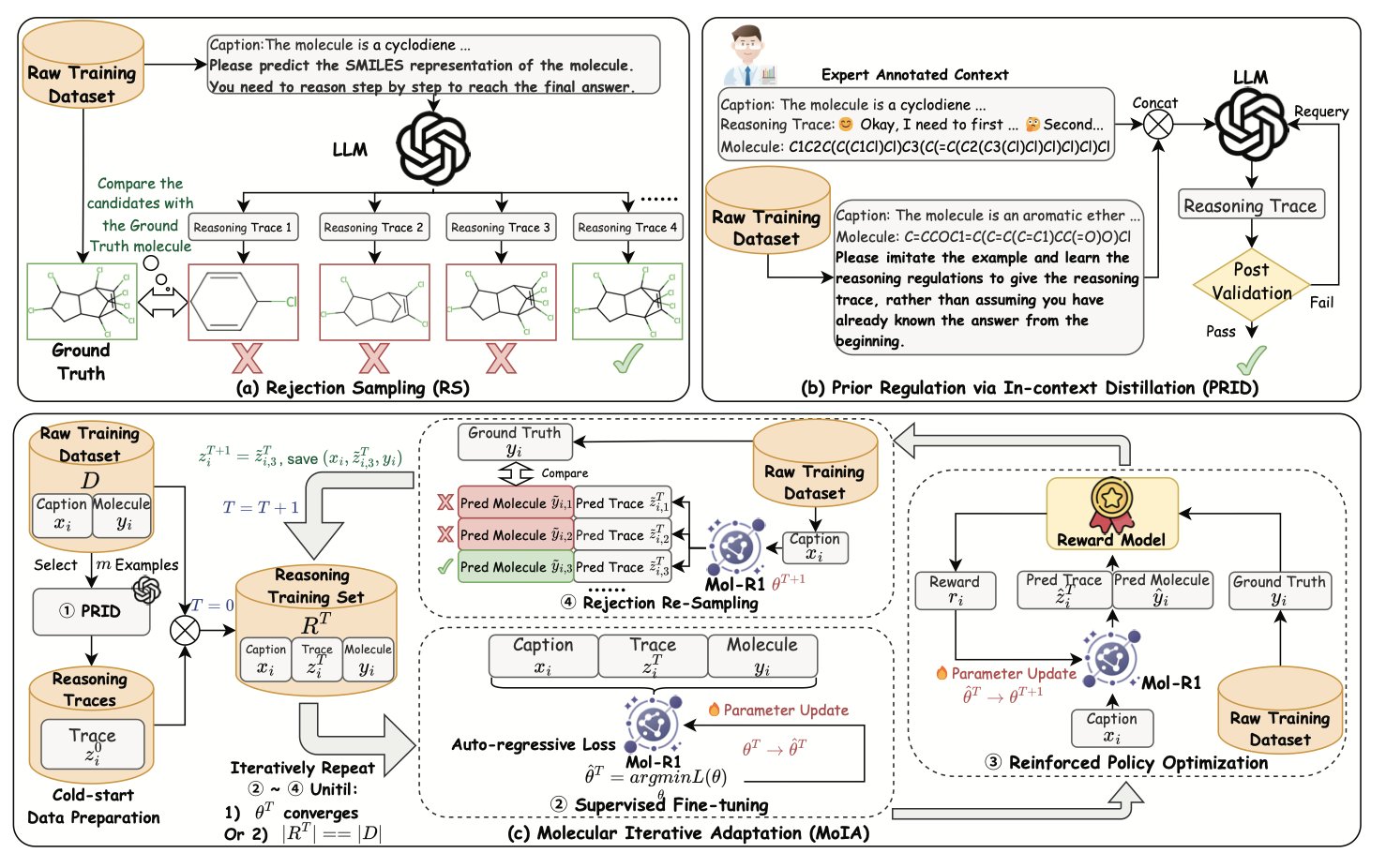
Mol-R1 的研究者想了个办法,叫「通过上下文蒸馏的先验调节」(Prior Regulation via In-context Distillation, PRID)。这就像给那个天才学生看了一页顶尖名师写的、步骤清晰的解题范例,然后跟他说:「看懂没?照这个逻辑和风格,给我出 100 道类似的题,再把解题过程写得一样漂亮。」
通过这种「以点带面」的蒸馏,他们用极少的专家标注,就引导 LLM 自己生成了一本厚厚的、高质量的、充满详细推理步骤的「化学思考题集」,解决了「没有教科书」这个根本问题。
光读书不够,还得刷题。
有了教科书,下一步就是怎么高效学习。Mol-R1 用了一套叫「分子迭代适应」(Molecule Iterative Adaptation, MoIA)的训练策略,完美模拟了学霸的学习路径:
1. 监督微调(Supervised Fine-Tuning, SFT) :这好比学生认真「背诵」我们刚印的教科书,学习所有正确范例,模仿专家的思维方式。
2. 强化策略优化(Reinforcement Policy Optimization, RPO) :这好比老师开始出一些书上没有的新题,让他自己「练习和试错」。做对了给高分(奖励),做错了给低分。通过这种反馈,他会慢慢自主探索教科书没覆盖到的、更广阔的化学空间。
MoIA 最关键的地方在于,它是一个迭代循环。学生读完一遍书,就去做一套题;做完题带着新感悟,再回去读一遍书;然后再去做一套更难的题……通过这种「学习 - 练习 - 再学习」的循环,模型的能力持续螺旋式上升。
最终结果:一个可以信任的「学霸」
经过这番「特训」,Mol-R1 不仅在生成正确分子的能力上超越了现有模型,它生成的那些「解题步骤」,也变得前所未有的清晰、连贯且符合化学逻辑。
这项工作还有一个惊喜的发现:AI 写出的「解题步骤」质量越高,最终答案就越靠谱。 这就像一个能把每一步都讲得明明白白的学生,他最后算对的概率,远比那个只会给你一个光秃秃数字的同学要高。
📜Title: Mol-R1: Towards Explicit Long-CoT Reasoning in Molecule Discovery
📜Paper: https://arxiv.org/abs/2508.08401
4. PEPBI 数据库:连接蛋白 - 多肽结构与热力学的桥梁
用 AI 设计多肽药物,是现在最火的赛道之一。但大家都有一个共同的难处:能拿来训练 AI 的好数据,实在太少。
缺的不是一般数据,而是一种「情侣套餐」:一边是高清的蛋白 - 多肽复合物三维结构,另一边是与之配对的、实验室测出来的精确结合热力学数据。
结构,就像一张照片,告诉我们多肽是怎么结合的。
热力学数据(吉布斯自由能ΔG,焓ΔH,熵ΔS),则像一份体检报告,告诉我们它俩结合得有多好,以及驱动这场结合的到底是「激情」(焓驱动)还是「合适」(熵驱动)。
只有把结构和能量这两样东西凑齐,AI 才能真正学懂蛋白 - 多肽相互作用的「底层逻辑」,而不是只会「看图说话」,死记硬背几何形状。这样,模型才能开窍,明白为什么有时候形成一个氢键(焓驱动),比填满一个疏水口袋(熵驱动)更关键。
这篇发表在Scientific Data上的 PEPBI 数据库,就是来填这个大坑的。
PEPBI 的「含金量」在哪里?
- 筛选标准,堪比选秀 :数据库收录数据门槛很高,对入选的复合物有几条硬性规定:
- 多肽长度限制在 5-20 个氨基酸,这是治疗性多肽的「黄金尺寸」。
- 晶体结构分辨率必须好于 2.0 Å,保证每个原子的位置都清清楚楚。
- 复合物之间的序列相似度不能太高,确保数据「不撞脸」,避免 AI 模型「偏科」。
- 多肽长度限制在 5-20 个氨基酸,这是治疗性多肽的「黄金尺寸」。
- 热力学数据,金标准认证 :库里的ΔG, ΔH, ΔS 值,都来自等温滴定量热法 (Isothermal Titration Calorimetry, ITC) 这类「金标准」实验,数据可靠。
- 附赠「特征大礼包」 :数据库不只提供「原材料」。研究者们还用 Rosetta 等工具,给每个复合物预先算好了 40 种界面性质,比如界面埋藏面积、氢键数量、形状互补性等。
这就好比,数据库不仅给了你「食材」,还顺手把「菜」都洗好切好了。搞 AI 建模的研究者可以直接端走这些特征来训练模型,省去了大量数据预处理的功夫。
PEPBI 数据库的价值
📜Paper: https://www.nature.com/articles/s41597-025-05754-7
5. rbiol:用「虚拟细胞」训练更懂生物学的 AI
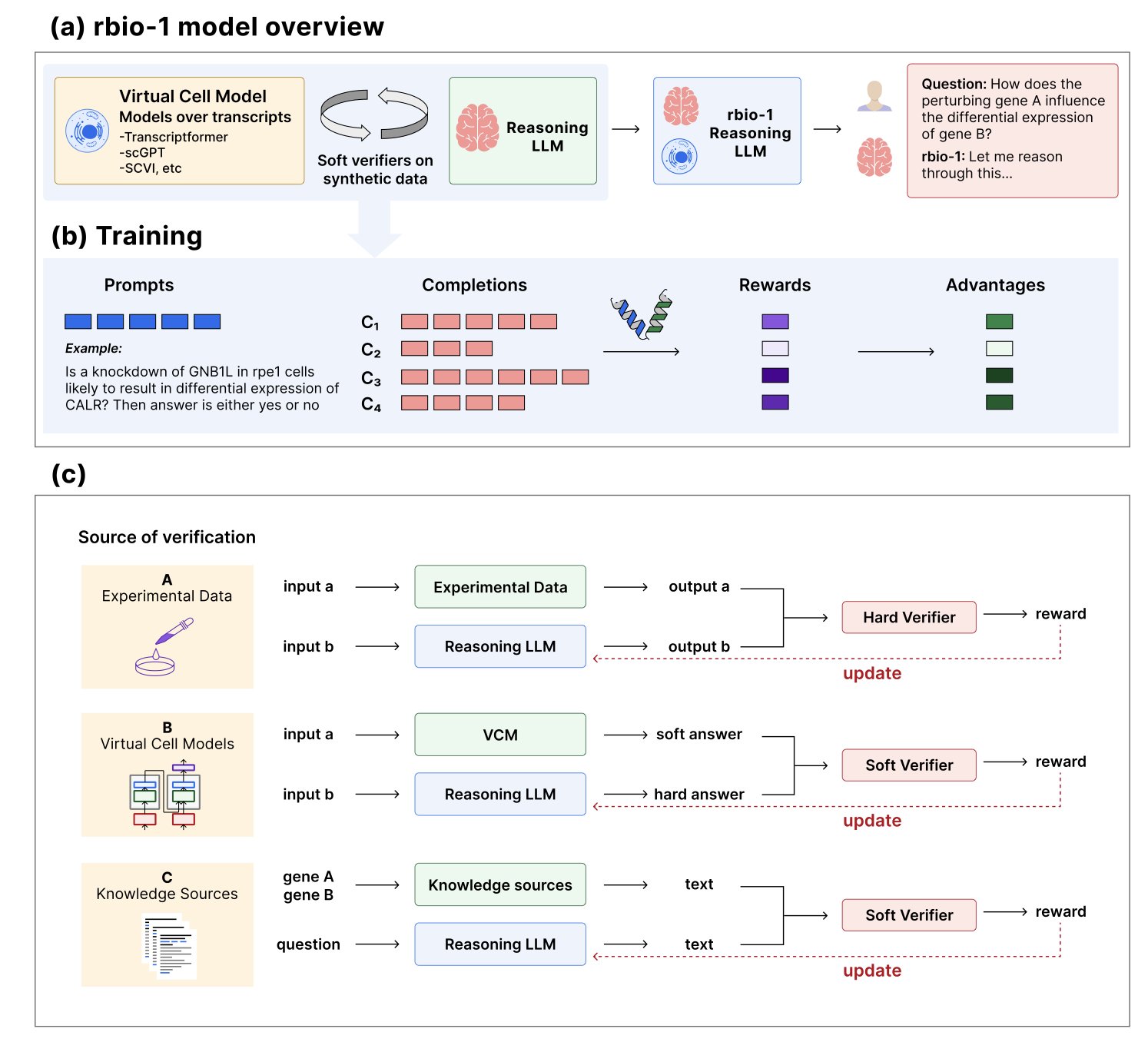
训练生物学 AI,就像带医学生。最好的方法当然是让他亲自下场做实验,从一手数据里找感觉。但生物实验这东西,出了名的烧钱还耗时,总不能为每个问题都砸钱做个大规模实验吧?经费在燃烧,头发在减少。
所以,能不能找个便宜点的法子?rbio 模型就想到了一个主意:请个「虚拟老师」来给 AI 上课。
什么是「虚拟老师」和「软验证」?
这个「虚拟老师」,其实是现成的各种生物学世界模型(biological world models)。这些模型虽不完美,但已经内化了大量人类已知的生物学知识和规律,就像一位老教授,肚子里都是货。比如,有的模型就能预测,给细胞敲掉某个基因后,它的转录组会怎么天翻地覆。
论文提出的「软验证」(soft verification)范式,就是让一个通用的大语言模型(LLM),拜这位「虚拟老师」为师。
训练过程是这样的:
1. 先向 LLM 提问,比如:「如果我把 A 基因干掉,B 基因的表达量会增加还是减少?」
2. LLM 给出自己的推理和答案。
3. 然后把同一个问题,丢给「虚拟老师」(比如某个转录组预测模型),让它也算一遍。
4. 最后,把「虚拟老师」的答案当作标准答案(软标签),去批改 LLM 的作业,告诉它哪里答错了、该怎么改。
经过成千上万轮这样的模拟问答和随堂测验,LLM 就逐渐把「虚拟老师」肚子里的生物学知识,「蒸馏」进了自己的脑子里。
这套方法管用吗?
结果有点惊人。用这种省钱方式训练出来的 rbio 模型,去预测真实世界的基因扰动实验时,表现竟然和那些拿真实实验数据喂大的模型不相上下,在某些任务上甚至还反超了。
这说明知识确实可以被高效传递和「蒸馏」。LLM 不只是在死记硬背模拟器的输出,它似乎真的悟到了一些底层的、能举一反三的生物学推理能力。
「软验证」的老师来源也可以很广。我们可以同时请来好几位不同领域的「虚拟老师」——一位精通转录组,一位专攻基因本体论,再来一位擅长蛋白质相互作用。让 LLM 博采众长,最终成为一个全面发展的「学霸」。
这项工作为构建通用的「虚拟细胞」推理引擎,画出了一条清晰可行的路线图。它告诉我们,不必等到把细胞里的所有秘密都靠实验挖出来,才动手构建有用的 AI。我们可以先把现有的知识,固化成一个个「世界模型」,然后用这些模型,去教出更聪明、能与我们对话、帮我们思考的下一代生物学 AI。
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.18.670981v3