
目录
- SmilesT5 通过专为化学家设计的预训练任务,证明了在分子语言模型中,“更聪明”的训练远比“更大”的数据集更有效。
- 这篇 Nature 论文展示了一个由 AI 智能体主导、人类专家指导的“虚拟实验室”框架,它不仅成功设计出针对新冠新变种的纳米抗体,更预示着一种全新的、可能颠覆传统研发协作模式的科研范式。
- AlphaFold3 在预测能折叠成两种构象的蛋白质时,常常会把两种结构硬塞在一起,生成一个物理上不可能存在的“缝合怪”,暴露出其处理序列模糊性的根本局限。
- SKiM-GPT 巧妙地将传统的文献共现搜索与大语言模型(LLM)结合,为海量生物医学假说的快速筛选和验证,提供了一套出乎意料的、脚踏实地的解决方案。
- 仅凭氨基酸序列预测蛋白质相互作用(PPI)正从一个计算领域的幻想,迅速变为药物发现的实用工具,而深度学习,尤其是 Transformer 模型,正在彻底改变我们筛选靶点和设计生物药的方式。
1. SmilesT5:AI 制药,告别暴力美学
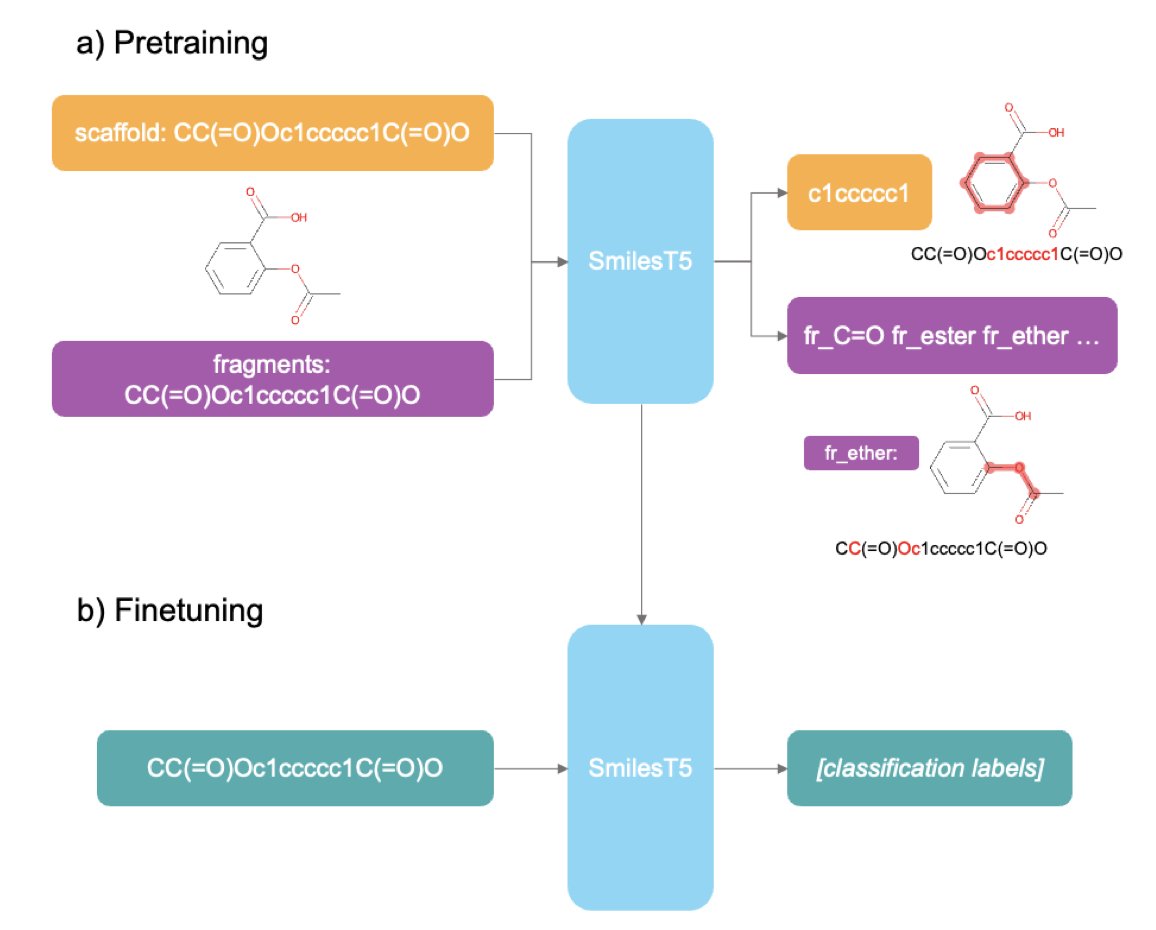
说实话,现在看到“新型语言模型”这几个字,第一反应是我们真的还需要另一个用几亿个分子暴力堆出来的大家伙吗?这些模型就像是装备了核武器的婴儿,力量巨大,但对世界的理解却粗糙得可笑。
但 Hothouse Therapeutics 这篇关于 SmilesT5 的论文没有走“大力出奇迹”的老路,而是问了一个更根本的问题:我们该怎么教 AI“说人话”——不,是“说化学家的话”?
传统的 Masked Language Modeling,就像把一本有机化学教科书撕碎了,随机盖住几个字,让一个外行去猜。他或许能学会一些基本的单词搭配,但对化学反应的机理、分子骨架的意义可能一无所知。SmilesT5 的玩法就高明多了,它的预训练任务非常“内行”。
比如,它有一个任务是“重建 Murcko 骨架”。你给它一个完整的分子 SMILES,让它反向预测出这个分子的核心骨架。这简直太妙了!这不就是在训练 AI 的“药化直觉”吗?一个经验丰富的化学家,拿到一个新分子,第一眼就是看它的核心环系和骨架。这决定了分子的基本构象、理化性质和潜在的靶点亲和力。SmilesT5 在预训练阶段就在干这个,它学的不是零散的原子连接,而是分子的“灵魂”。
另一个任务是从 SMILES 中识别分子片段。这不就是我们化学家每天在做的事吗?把一个复杂的分子拆解成我们熟悉的砌块——苯环、酰胺、哌嗪——来分析它的合成路线和 SAR(构效关系)。通过这种方式训练,模型自然就理解了哪些片段是常见的“优势骨架”,哪些是需要警惕的“毒性基团”。
让我眼前一亮的是它的数据效率。只用了区区 100 万个分子就完成了预训练,效果居然不输那些动辄吞掉几千万甚至上亿数据的模型。这说明什么?说明“教得好”比“喂得多”重要得多。这对于没有谷歌、英伟达那种算力资源的团队来说,简直是天大的好消息。你不需要一个超算中心,在自己的工作站上也能训练出一个能打的模型。
模型的应用方式也很接地气。如果你有资源,当然可以对整个模型进行微调,让它专攻你的特定任务,比如预测 hERG 抑制或者肝毒性。但如果你的计算资源有限,也没关系。你可以把 SmilesT5 当成一个“特征提取器”,把它为每个分子生成的“嵌入”(embeddings)拿出来,然后用这些特征去跑你熟悉的 XGBoost 或者随机森林。计算开销小得多,但因为这些嵌入已经蕴含了丰富的化学知识,效果可能出奇地好。这大大降低了 AI 辅助药物发现的门槛。
📜Title: SmilesT5: Domain-specific pretraining for molecular language models
📜Paper: https://arxiv.org/abs/2507.22514v1
💻Code: https://github.com/hothousetx/smiles_t5
2. AI 智能体团队:药物发现的下一个“老板”?
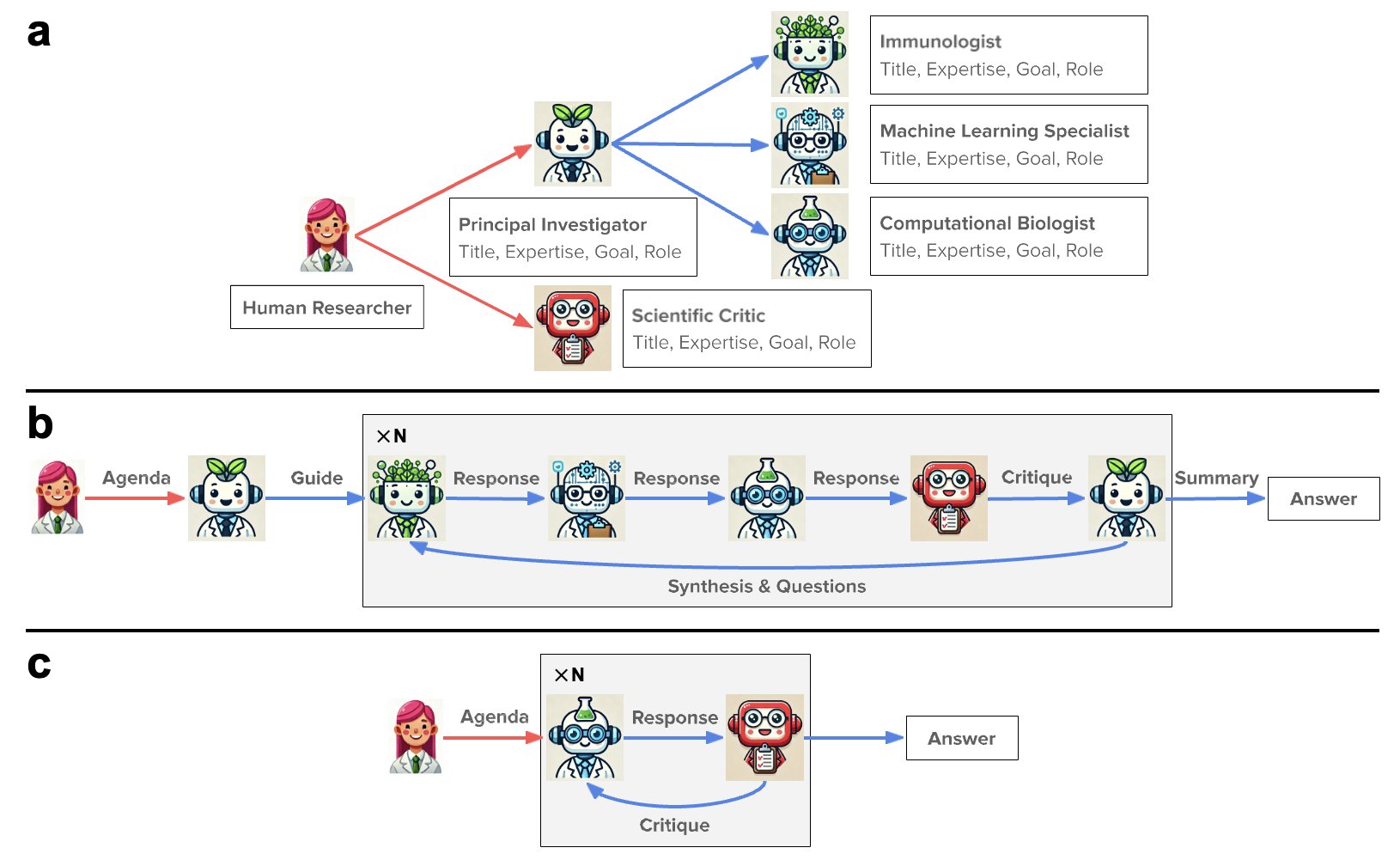
好了,这篇 Nature 论文读起来就像是科幻小说照进了现实。它讲的不是又一个预测蛋白结构的“黑科技”算法,而是组建了一个由 AI 驱动的“虚拟实验室”,让一群 AI 智能体(Agents)自己开会、分工、吵架,最后设计出了能中和最新新冠变种的纳米抗体。这事儿可比单纯跑个模型要野多了。
我们都知道,一个项目最头疼的是什么?是沟通。
生物学家、化学家、计算科学家,每个人说的都是“行话”,常常鸡同鸭讲。一个简单的想法,来回拉扯几周是家常便饭。这篇论文的作者们似乎想从根子上解决这个问题。他们搞了个大语言模型(LLM)当“总指挥”,然后给它配了一帮“AI 专家”手下。
这个团队里有负责提需求的“项目负责人 AI”,有懂蛋白设计的“生物学家 AI”,还有专门跑模拟的“计算专家 AI”。
人类研究员干嘛呢?当个甩手掌柜,提出一个最高指令:“嘿,给我设计点能干掉最新新冠病毒的纳米抗体。”
接下来,这群 AI 就开始自己忙活了。它们会调用各种我们熟悉的“重型武器”,比如用 ESMFold 和 AlphaFold-Multimer 预测结构,用 Rosetta 做对接和优化。
这就像什么?就像你雇了一个超级聪明的项目经理,他不仅能听懂你的战略意图,还能自己去招募和管理一堆顶尖技术专家,你只需要等着看结果就行。整个过程,人类的角色从“动手干活”变成了“定义问题”和“最终拍板”。
我们来看看它们交出的成绩单。92 个全新的纳米抗体设计,这不算什么。关键是,实验验证下来,超过 90% 的蛋白都能顺利表达并保持可溶。做过蛋白表达的人都知道这是个什么概念——这可不是一个可以忽略的成功率,很多博士生的课题可能就卡在蛋白不表达或者一表达就沉淀上了。这说明 AI 设计出来的东西,至少在“可实现性”上是靠谱的。更妙的是,其中两个候选分子表现出了独特的结合模式,意味着 AI 可能发现了一些人类专家容易忽略的角落。
当然,看到这种东西,我脑子里立刻会冒出一百个问题。这些设计的亲和力到底多强?KD 值是多少?除了结合新冠,它们会不会乱碰体内其它蛋白,搞出脱靶毒性?成药性可不只是溶解度好就完事了,稳定性、半衰期、免疫原性呢?这些都是从一个漂亮的计算模型到一个真正的药物之间,隔着的万水千山。论文里没细说,这也很正常,毕竟这只是一个概念验证。
但这篇论文的真正价值在于展示了一种全新的研发“组织形式”。
过去,我们说 AI 辅助药物发现,AI 是个工具,像个高级计算器。现在,AI 成了一个“合作者”,甚至是一个“团队领导”。它把不同学科的知识和工具串联了起来,让一个原本需要多人团队耗费数月才能完成的复杂工作流,被压缩在了一个人机交互的框架里。
这意味着什么?一个优秀的生物学家,即便对计算化学一知半解,未来也可能通过指挥这样一个 AI 团队,独立完成早期药物分子的设计和筛选。跨学科的壁垒,正在被 AI 用一种我们没想到的方式夷为平地。
所以,这会让我们失业吗?短期内不会。AI 团队还需要人类来提出那个“伟大的问题”。但我们的工作方式,恐怕要变了。以后组建项目团队,除了要找对人,可能还要考虑该用哪个“AI 团队”了。你的下一个项目合作者,可能真的没有博士学位,但它肚子里装着整个互联网的知识,而且 7x24 小时待命。
📜Title: The Virtual Lab of AI agents designs new SARS-CoV-2 nanobodies
📜Paper: https://www.nature.com/articles/s41586-025-09442-9
3. AlphaFold3 翻车:当 AI 遇上“双重人格”的蛋白质
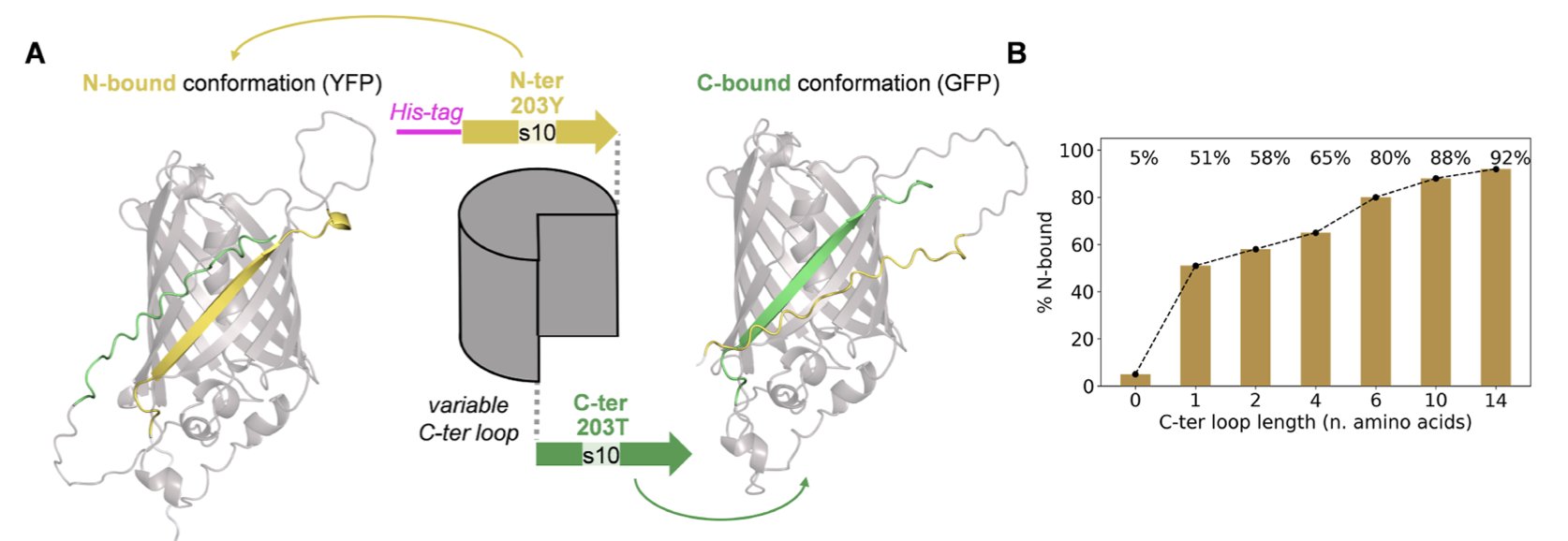
AlphaFold3 无疑是结构生物学领域的“新神”,但当我们不再喂给它那些安分的、只有一种稳定构象的蛋白质,而是扔过去一个真正的“刁钻”问题——一个被设计成可以折叠成两种不同结构的蛋白质时,会发生什么?这篇新研究就干了这事,结果相当有意思,也给搞研发的人泼了一盆恰到好处的冷水。
研究者们设计了一些所谓的“交替框架折叠”(alternate frame folding)系统。你可以把它想象成一个英文句子,通过移动空格的位置,可以读出两种完全不同的意思。这些蛋白质序列就是这样,它们内部的一段重复序列,可以有两种不同的配对折叠方式,从而形成两种截然不同的三维结构。这对于任何结构预测算法来说,都是终极考验。
结果呢?AlphaFold3 常常会“精神分裂”。
它没有像一个理智的科学家那样,在两种可能性中选择一个最合理的,而是试图把两种构象都表现出来。最终生成的模型,就是一个把两种结构强行叠加在一起的“缝合怪”。
你能看到 β-折叠链从一个本应是桶状结构的中间穿心而过,或者氨基酸侧链挤在一个物理上绝无可能的位置。这就像让 AI 同时画一只猫和一条狗,它不是画出一只“猫狗兽”,而是把猫和狗的线稿重叠在一起,得到一团无法辨认的混乱。
更有趣的是,这暴露了 AlphaFold3 和 AlphaFold2 的一个核心区别。用过 AF2 的人都知道,我们可以通过“魔改”多序列比对(MSA)来“诱骗”它,让它探索不同的构象。这就像给侦探提供不同的线索,他可能会推导出不同的案情。但 AF3 减少了对 MSA 的依赖,这把双刃剑在这里就割到了自己。它变得更像一个“黑箱”,你很难从外部去引导它。它看到了序列本身固有的模糊性,然后就卡住了,输出了一个代表着“犹豫”的错误结果。
论文还提到了一个做药的人在日常工作中经常会忽略的细节:His-tag。这个我们用来纯化蛋白质的“小尾巴”,因为它没有固定的结构,在 AF3 的预测里也成了麻烦制造者。模型会倾向于给这个柔性的尾巴强行安上一个结构,结果往往导致主蛋白区域出现不合理的空间碰撞。这再次提醒我们,AI 工具远没有“理解”生物化学的全部。你输入的每一个原子,都可能成为影响结果的变量。
研究者们还用基于钙调蛋白的构象开关验证了这一点,发现问题是普遍存在的,并非 GFP 独有的怪癖。
所以,AlphaFold3 是个强大的工具,但他也有它的“阿喀琉斯之踵”——那就是模糊性。
当一个序列本身就包含“向左走,还是向右走”的内在矛盾时,AF3 的现有架构就会短路。这项工作不是为了贬低 AF3,而是为了绘制出它的能力边界。对于试图设计新药、新蛋白或者理解疾病机理的人来说,清楚地知道工具的局限性,远比盲目相信它的能力要重要得多。这能让我们知道,什么时候可以信赖 AI,什么时候必须回归实验,用我们自己的化学直觉和瓶瓶罐罐来寻找答案。
📜Title: Structure Prediction of Alternate Frame Folding Systems with AlphaFold3
📜Paper: https://pubs.acs.org/doi/full/10.1021/acs.jcim.4c00420
4. 告别文献大海捞针:SKiM-GPT 智能验证假说
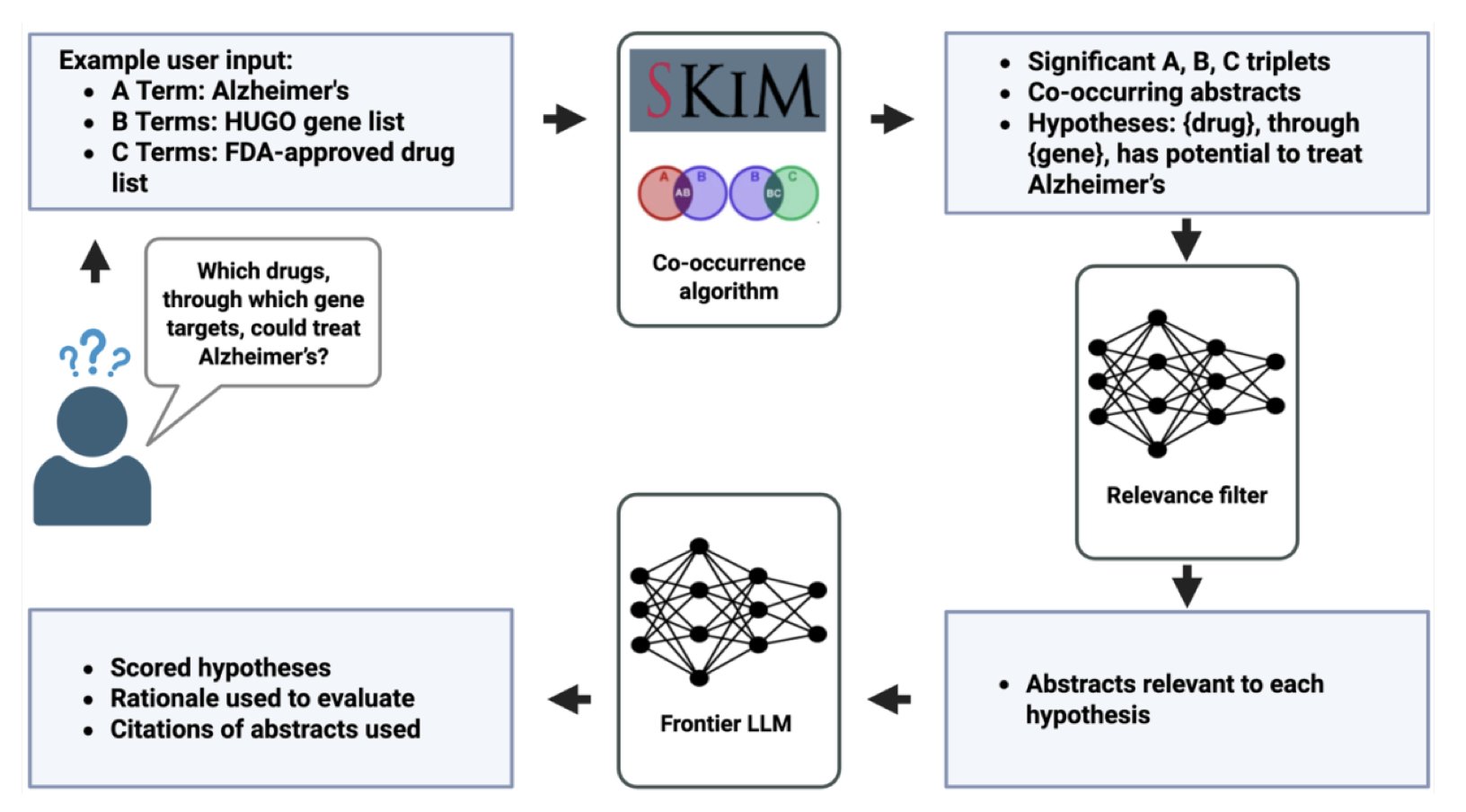
每天早上打开 PubMed,面对庞大的文献数量,我们最缺乏的无疑是时间。因此,各类“AI 赋能科研”的工具纷纷涌现,坦白说,大部分都只是空谈而已。它们要么是过于自信的“黑箱”,要么仅仅是将简单的关键词搜索包装而自称为“智能”。
SKiM-GPT 这套系统的聪明之处在于,它深刻理解了大语言模型(LLM)的优点和致命缺陷。
LLM 就像一个天赋异禀但有点疯癫的实习生,知识渊博,但如果你给它的任务太开放,它就很容易开始“自由发挥”,也就是我们常说的“一本正经地胡说八道”(hallucination)。你直接问它“某个基因和某个疾病有关系吗?”,它给你的答案可能一半是事实,一半是它根据语言模式脑补出来的剧情。
SKiM-GPT 整个流程分两步:
第一步,用一个叫 SKiM (Serial KinderMiner) 的工具进行海选。它儿没什么花哨的,它就是一个勤勤恳恳的“记账员”,在海量的 PubMed 文献摘要里,统计哪些术语(比如一个基因名和一个疾病名)同时出现的频率远高于随机概率。它不懂生物学,但它懂统计。这就像在成千上万份报告里,找出所有同时提到“失火”和“XX 大楼”的文档。这种方法虽然简单,但非常高效,能快速圈定一个高度相关的文献范围。
第二步,才是让 LLM 上场精读。这时候,LLM 的任务不再是漫无边际的“探索”,而是一个极其聚焦的任务:“嘿,这里有 20 篇我为你挑好的论文摘要,你仔细读读,然后告诉我,它们到底支不支持‘X 基因导致 Y 疾病’这个假说?”
你看,这才是驾驭 LLM 的正确姿势。你把它从一个“创意总监”变成了一个“高级尽职调查分析师”。在这样一个有明确边界和输入源的任务里,LLM 的幻觉问题被极大抑制,而它强大的文本理解和总结能力则能发挥得淋漓尽致。
结果呢?
在一个包含 14 个“疾病 - 基因 - 药物”假说的基准测试中,它的判断和人类专家的判断高度一致,Cohen’s kappa 分数达到了 0.84。在统计学上,这通常意味着“高度一致”。换句话说,这台机器的表现,几乎和你找另一个领域专家来做二次判断的结果差不多。
更关键的是,它不是一个傲慢的黑盒子。它会把打分、结论以及作为证据的论文摘要全都摆在你面前。你可以亲自检查,看看它的逻辑链是否站得住脚。这种“玻璃盒”式的设计,对于需要在实验室里用真金白银和宝贵时间去验证假说的人来说,是绝对的刚需。
当然,目前它主要啃的是论文摘要,我们都知道,真正的魔鬼细节往往藏在方法部分或者补充材料的某个角落里。让 AI 精准理解全文,尤其是图表和实验数据,是下一个挑战。而且,它的基础仍然是已发表的文献,如果文献本身就有偏见或者错误(这种情况可不少见),那它也只能是高质量地总结这些垃圾信息。
尽管如此,SKiM-GPT 没有吹嘘自己能取代科学家,而是老老实实地去解决一个最令人头痛的瓶颈问题:如何在信息洪流中快速、高效地验证一个新想法是否值得投入资源。
最聪明的 AI 应用,往往不是用最炫的技术去挑战最宏大的问题,而是用恰当的技术组合,去解决一个具体、真实且痛苦的难题。
📜Title: SKiM-GPT: Combining Biomedical Literature-Based Discovery with Large Language Model Hypothesis Evaluation
📜Paper: https://www.biorxiv.org/content/10.1101/2025.07.28.664797v1
💻Code: https://github.com/stewart-lab/skimgpt
5. AI 预测蛋白互作:新药发现的游戏规则改变者?
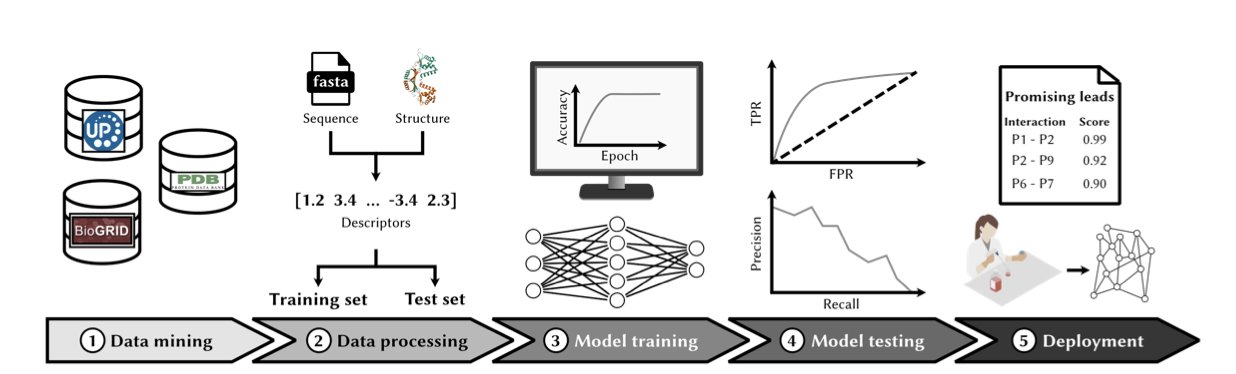
几十年来,开发针对蛋白质 - 蛋白质相互作用(PPI)的药物,一直有点像试图用手抓住青烟——充满希望,但又无比困难。PPI 的界面大多又大又平,对传统的小分子药物来说简直是噩梦。所以,我们越来越多地将目光投向了生物药。
但问题来了:茫茫多的 PPI 中,到底该打哪一个?设计生物药时,又该如何下手?
这时候,计算方法登场了。设想一下,你把两种蛋白质的氨基酸序列扔进一个黑匣子里,它就能告诉你它们俩“看对眼”的概率有多大。这听起来有点科幻,但我们离这个目标越来越近了。
过去的方法,更像是通过看简历来配对——找找序列或者结构域上有没有相似之处。有时候管用,但也就那样了。
现在是基于 Transformer 的深度学习模型,则完全是另一个层次。它们不再是简单地比对“简历”,而是试图去理解蛋白质这门独特的语言。模型通过学习海量的序列数据,掌握了决定蛋白质间“化学对话”的深层语法和潜规则,判断它们在物理上是否会发生勾连。
当然,最大的麻烦在于数据。我们现有的 PPI 数据库存在严重的“偏科”。关于 p53 这类明星分子的互作研究堆积如山,但对成千上万个“路人”蛋白,我们几乎一无所知。用这种数据去训练模型,就好比只给 AI 看猫和狗的照片,然后指望它能认识鸭嘴兽。这注定会碰壁。更别提那个老大难的类别不平衡问题了——毕竟,宇宙中绝大多数蛋白质组合是不会相互作用的。如果模型偷懒,永远只预测“不互作”,它也能获得高达 99% 的准确率,但这显然毫无用处。
那这些工具在现实中到底能干嘛?
首先是靶点发现。假设你手上有一个新的肿瘤靶点,想知道它在细胞里都和谁“勾搭”。与其苦哈哈地做上几个月的酵母双杂交或者免疫共沉淀,你现在可以在一个下午就完成对整个已知蛋白质组的计算筛选。这并不能取代湿实验,但它能给你一份按优先级排好的假说清单,让你能把宝贵的实验资源用在刀刃上。
更令人兴奋的是在生物药设计上的应用。你想设计一条多肽来阻断某个特定的 PPI?这些模型可以帮你快速迭代序列,筛选出那些最有可能结合的候选分子。这对于设计过程来说,是一个巨大的飞跃,让我们能够摆脱纯粹靠人海战术进行筛选的窘境。无论是设计治疗性多肽,还是优化抗体,这都为我们提供了一个强大的计算“副驾驶”。
这个领域的发展速度快得惊人。整个学界都在呼吁建立标准化的基准数据集,这事儿太对了。我们需要一个公平的竞技场,而不是各家都在自己的后院赛道上跑分。随着蛋白质语言模型(PLM)变得越来越聪明,它们预测蛋白质之间复杂“握手”的能力只会越来越强。
📜Title: Sequence-based protein-protein interaction prediction and its applications in drug discovery
📜Paper: https://arxiv.org/abs/2507.19805