
目录
- 暴力采样能提升 AlphaFold 对复杂蛋白界面的预测精度,但如何从海量模型中挑出最佳答案,仍是关键挑战。
- LightChem 将大语言模型的推理能力与成熟的化学计算工具相结合,为化学家提供了一个轻量、精准、且专业的 AI 助手。
- 研究者利用蛋白质结合位点的水分子结构作为「路标」,开发出一种引导式对接新方法,解决了柔性糖链分子的对接难题。
1. AlphaFold 大力出奇迹?暴力采样如何搞定蛋白复合物
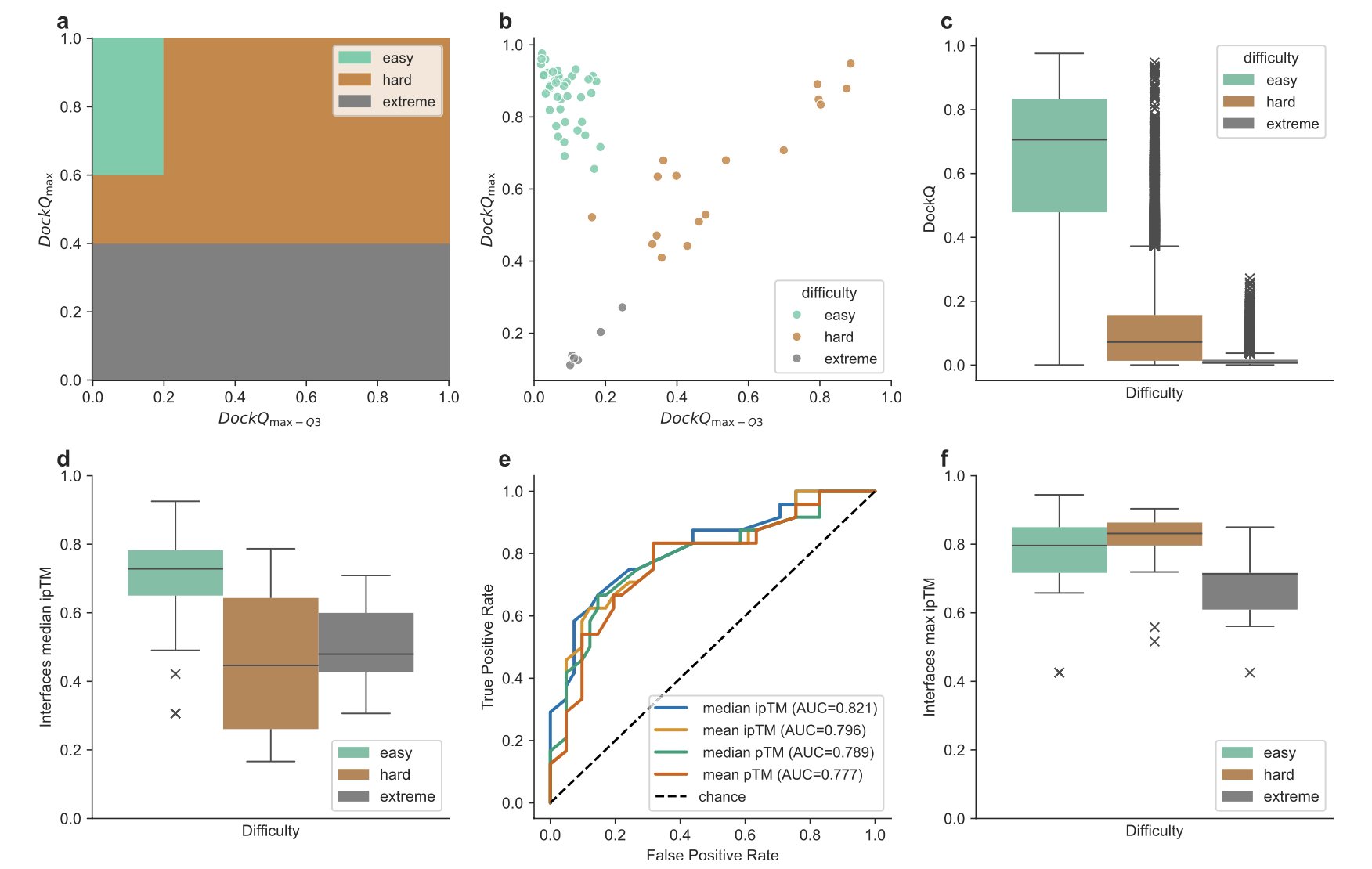
AlphaFold2 是一个突破性的工具,其预测单个蛋白结构的能力已很惊人。但将其用于蛋白 - 蛋白相互作用(Protein-Protein Interactions, PPIs),特别是预测两个蛋白如何结合时,它有时会出错。如同一个顶尖学生,基础题全对,但一遇到复杂的综合题,就可能给不出确切答案。
标准的 AlphaFold 运行一次,通常会提供 5 个模型。如果这 5 个模型看起来都不太对,或相互之间差别很大,基本就无计可施了。对于那些结合界面复杂、柔性大的「困难」靶点,这种情况很常见。
大力真的能出奇迹
这篇文章的研究者想了一个简单直接的办法:如果 5 个模型不够,那就生成 8000 个。这就是所谓的「暴力采样」(Massive Sampling)。
这个思路很直接。AlphaFold 在探索构象空间时,本身带有随机性。多跑几次,就等于给了它更多机会「撞」到正确的构象。这如同在沙滩上找一粒特定的沙子,用手抓 5 次可能找不到,但若开来一辆卡车,将整片区域的沙子都筛一遍,找到的概率就大大增加了。
研究者们在 CASP16-CAPRI 竞赛上系统地测试了这个方法。他们将蛋白复合物的界面按预测难度分为「简单」、「困难」和「极端」三类。结果显示,对于「困难」级别的界面,暴力采样几乎总能生成高质量的结构模型,效果提升明显。
「偷懒」方法
为每个蛋白都生成 8000 个模型,计算成本巨大,需要动用超级计算机集群。
所以,这项工作最巧妙的地方,是他们找到了一个判断「是否需要采用暴力采样」的指标。
他们先用标准模式运行一遍 AlphaFold,得到几个模型,然后计算预测界面 TM 分数(interface predicted TM score, ipTM)的中位数。ipTM 分数可以理解为 AlphaFold 对自己预测的界面区域的自信程度。如果这个中位数很低,就说明 AlphaFold 自己也很「心虚」,感觉这个界面很难处理。
这就是一个信号。一旦出现这个信号,就果断启动暴力采样模式。
这个方法很实用。它帮助将计算资源用在刀刃上,避免了在那些本就简单的靶点上浪费算力。根据他们的数据,采用这种策略性采样,可以将预测总数从 8040 个模型减少到 2475 个,同时预测精度几乎没有损失。
新的挑战:大海捞针
暴力采样解决了一个问题,也带来了另一个问题。
现在你手里有 8000 个模型,其中可能藏着最接近真实结构的「金标准」。但你怎么把它找出来?
AlphaFold 自带的模型排序打分,在这种情况下不那么灵了。从 8000 个高度相似的候选中挑出最好的那一个,对现有的打分函数(Scoring function)是个巨大挑战。
研究者们也坦诚,他们没有解决这个问题。但他们做了一件更有价值的事:将所有数据,包括全部模型、AlphaFold 的打分以及竞赛官方的评估结果,全部公开。
这对整个领域来说是个宝贵的资源。它提供了一个绝佳的训练场和测试集,让所有从事算法开发的人,都能来尝试开发更精准的打分函数,解决这个「大海捞针」的问题。
对于研发人员,这项工作提供了一个处理棘手 PPI 靶点的新思路。当你遇到一个高价值但 AlphaFold 预测不佳的复合物时,可以考虑投入计算资源进行暴力采样,这可能会带来突破。同时,它也指明了下一个技术突破点——开发出能从海量数据中精准识别最佳模型的打分算法。
📜Title: MassiveFold Data for CASP16-CAPRI: A Systematic Massive Sampling Experiment
📜Paper: https://onlinelibrary.wiley.com/doi/10.1002/prot.70040
2. LightChem: 更懂化学的轻量级 AI,精准预测分子性质
LLM 知识面广,但让它设计药物分子或规划合成路线,结果常有谬误。这如同让一个通才去做高度专精的外科手术,风险很高。化学是一门精确的学科,差之毫厘,谬以千里。
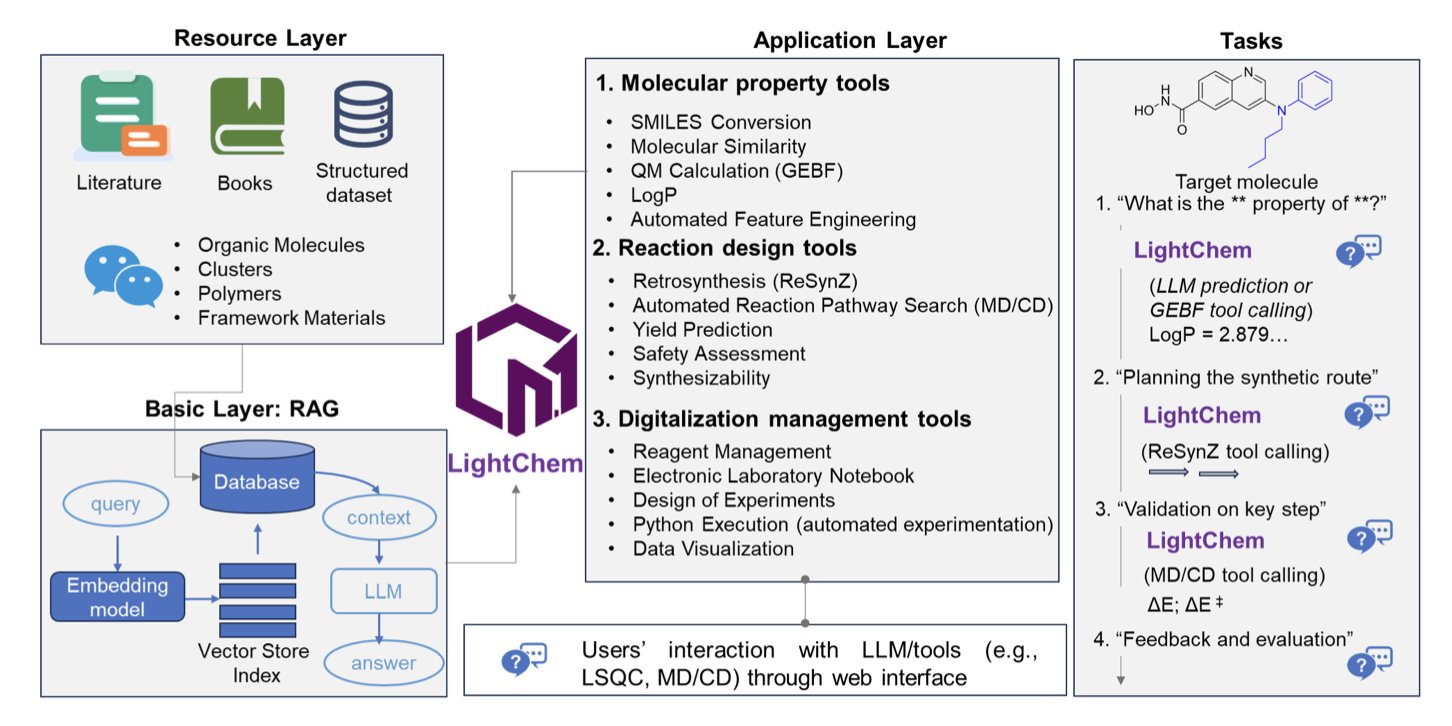
LightChem 的作者采用了另一条思路。他们将 LightChem 设计成一个专业的「项目经理」,它自己不掌握所有化学细节,但知道去哪里查资料(检索增强生成),以及该调用哪位「专家」(专业工具)来解决具体问题。
这个架构设计是整个工作的核心。当用户提出一个化学问题,比如预测分子的油水分配系数(PoLogP),LightChem 不会基于看过的海量文本去「创作」答案。它会启动一个专门用于此项计算的成熟模块,如 PoLogP 预测工具,然后将精确计算的结果用自然语言呈现给你。
这从根本上解决了通用大模型在专业领域容易出错的问题。对于研发人员来说,可靠性是第一位的。
它集成的「专家团队」包括:用于逆合成规划的 ReSynZ 模块,以及用于高精度计算的 CIM 和 GEBF 等量子化学软件包。LightChem 的能力建立在这些已被化学界广泛验证的计算方法之上。它像一个经验丰富的科学家,背后有一个装备精良的计算化学团队。
研究者用几个案例展示了其能力。预测候选药物的理化性质、设计合成路线,这些都是药物化学家的日常工作。LightChem 都能完成。它还能处理更大规模的计算,比如模拟分子在沸石团簇中的结合能,或绿色荧光蛋白(GFP)发色团的激发能,结果与实验值吻合。
当然,它并非万能。作者坦诚指出,在预测复杂聚集体的最高占据分子轨道 - 最低未占分子轨道(HOMO-LUMO)能隙和光学性质时,模型表现仍有待提高。这种坦诚划定了工具的能力边界,让我们知道在何种情况下可以信任它,何时需要保持谨慎。
最后,这个工具提供了一个网络界面,将复杂算法打包,降低了使用门槛。它不仅是一个理论模型,更像一个可以集成到实验室工作流里的实用助手。它把知识驱动的推理和第一性原理计算连接了起来。
📜Title: LightChem: A Lightweight Domain-Specific Language Model for Molecular Property and Reaction Prediction in Chemistry
📜Paper: https://doi.org/10.26434/chemrxiv-2025-wd80w
3. 水分子做向导,破解糖基对接难题
分子对接(molecular docking)中,处理柔性小分子已很复杂,而对接糖链(glycan)则更为困难。糖链分子大而柔韧,构象繁多。其在蛋白上的结合位点通常又浅又平,富含亲水基团,缺乏传统药物靶点的清晰「口袋」。这导致传统的对接软件常给出能量看似合理、但化学上错误的构象。
这项研究提供了一个新的解决思路。研究者关注一个问题:在糖链结合前,蛋白结合位点里有什么?
答案是水分子。这些水分子并非随机存在,它们与蛋白表面的氨基酸形成了稳定的氢键网络。
研究者意识到,这些水分子的位置,实际上标出了一张「相互作用热点图」。一个水分子能在此处与蛋白形成氢键,那么配体上一个合适的基团(如羟基)置于此位,也能形成类似的有利相互作用。
基于此,他们开发了一套名为「WII 引导方法」(WIIGA)的对接流程。
它的工作原理如下:
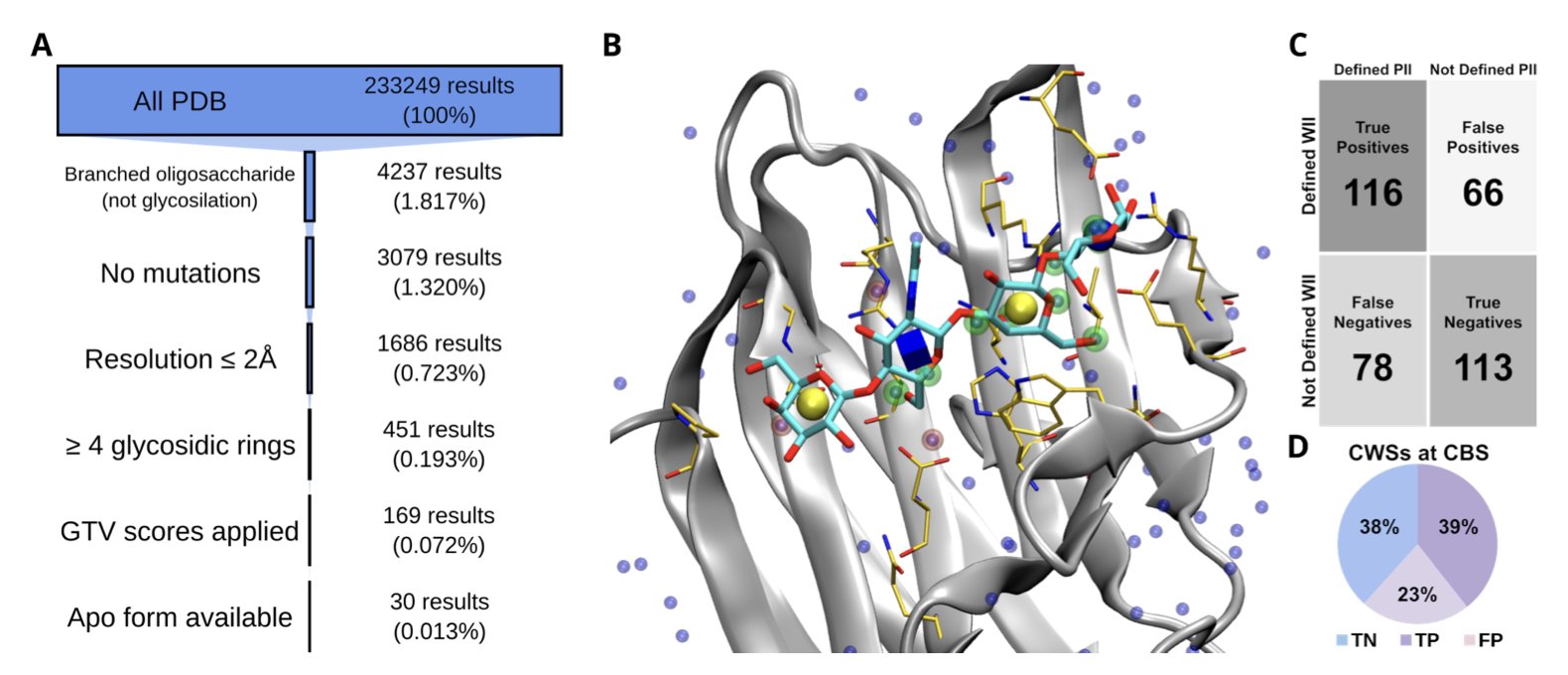
1. 首先,使用一个未结合配体的 apo 蛋白结构。
2. 识别结合位点内的所有水分子,将其位置定义为「水的理想相互作用位点」(Waters Ideal Interactions, WII)。
3. 然后,在使用 AutoDock Vina 进行对接计算时,为打分函数增加一个「奖励项」:若糖链配体上的某个原子,恰好落在某个 WII 热点区域,并能形成化学上合理的相互作用(如氢键),则该对接构象会获得加分。
这如同在寻宝游戏中提供一张标有红叉的地图,缩小了搜索范围,避免程序在无意义的构象空间中搜索。
为验证该方法,研究者们建立了一个包含 30 个高质量蛋白 - 寡糖复合物的测试集。结果显示,WIIGA 的表现全面优于几个主流的糖基对接工具,包括标准版的 AutoDock Vina、Vina Carb (VC) 和 GlycoTorch Vina (GTV),能更准确地预测出接近晶体结构的正确结合模式。
该方法的一个优点是,它仅需 apo 蛋白的结构。在药物研发中,通常更容易获得靶点蛋白自身的结构,而非其与配体结合后的复合物结构。WIIGA 在更接近真实场景的交叉对接测试中,依然表现稳健。
更有价值的是,此方法同样适用于药物化学家更关心的类药小分子——糖模拟物(glycomimetics),可用于设计靶向凝集素(lectin)等糖结合蛋白的小分子药物。
当然,该方法也有其局限。它的一个主要限制在于蛋白质本身的构象。若蛋白在结合配体时自身会发生剧烈的构象变化(即「诱导契合」),那么基于刚性 apo 结构的 WII 热点图可能就不准确。测试结果也证实了这一点:使用 apo 结构或 AlphaFold3 预测的模型,其对接准确性低于使用实验测定的 holo 结构。这提示,对于构象柔性大的靶点,单一晶体结构可能不够,或需借助分子动力学模拟或结构系综来获得更全面的信息。
这项工作为解决糖基对接这一难题提供了一个实用且巧妙的工具,它也说明,有时解决复杂问题的关键,就藏在像水这样最常见、最易被忽略的细节之中。
📜Title: Guided docking using solvent structure information improves the prediction of protein-glycans complexes
📜Paper: https://doi.org/10.26434/chemrxiv-2025-8qvgh