
目录
- CellForge 不只是一个预测工具,它是一条能自动将生物学问题转化为高效计算模型的「流水线」,有望将科学家从繁琐的建模工作中解放出来。
- AI 模型给蛋白质口袋画了一张「倾向图」,告诉药物化学家把不同的原子放在哪里才能获得最佳结合。
- 这个模型终于开始正视药物发现中那个被长期忽视的「房间里的大象」——溶剂效应,强迫 AI 承认分子是活在「水」里,而不是真空中。
1. CellForge:AI 打造的虚拟细胞流水线
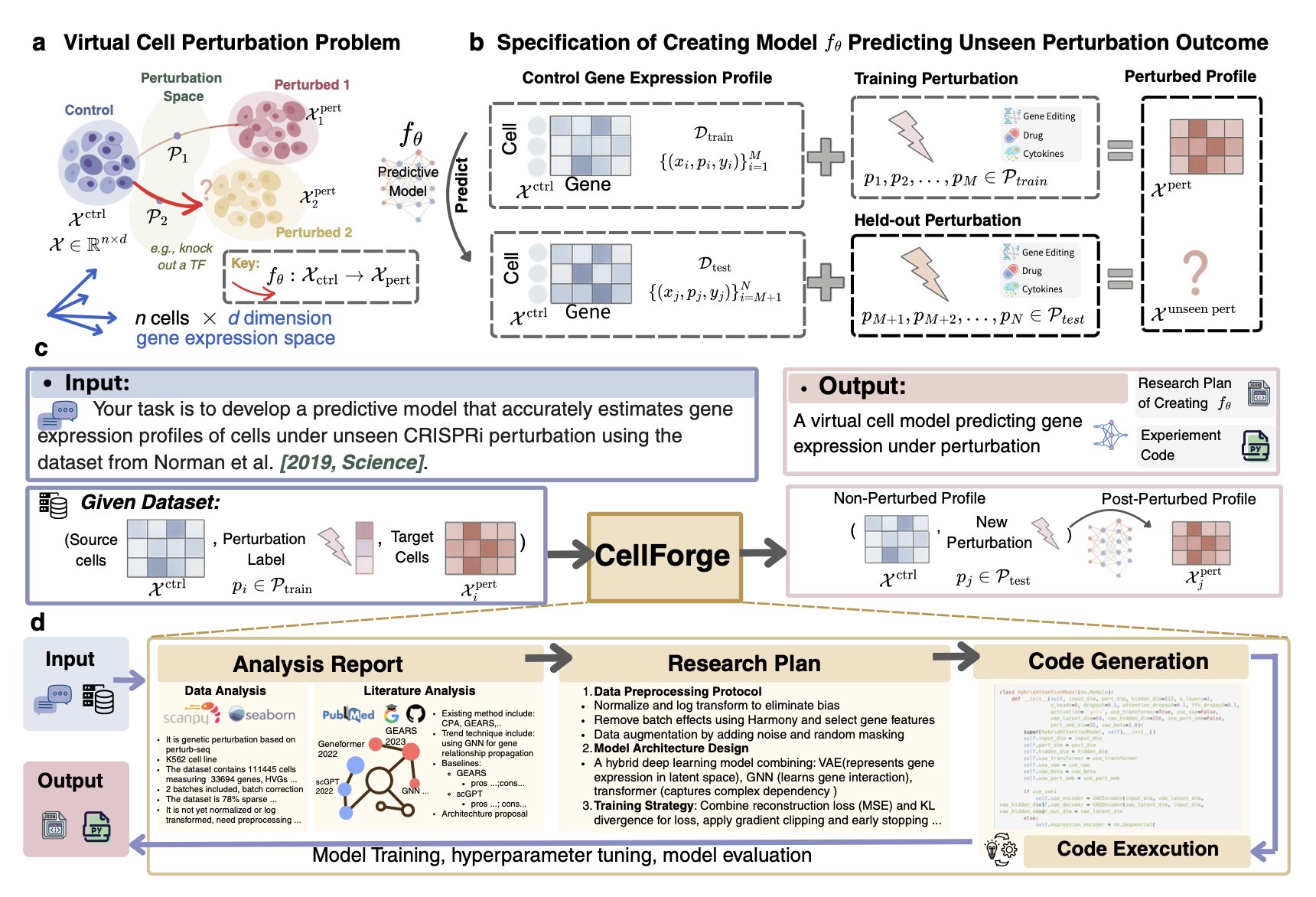
计算生物学领域梦想着能有一个「虚拟细胞」——一个可以在电脑里完美模拟真实细胞行为的模型。但现实是,搭建这样一个模型的过程痛苦不堪。你需要一个既懂生物学,又懂机器学习,还能熟练编程的「独角兽」科学家,花上几个月甚至更久的时间,在无数种模型架构和参数中苦苦挣扎。
现在,来自 Mark Gerstein 和 Fabian Theis(这两个名字在圈内分量可不轻)等大佬实验室的 CellForge,似乎想彻底终结这种手工作坊式的建模方式。
CellForge 干的不是小修小补,它直接构建了一条全自动的「虚拟细胞模型生产线」。这套系统的野心极大,它试图包揽从头到尾的所有工作。你只需要告诉它你的科学问题(比如,我想知道某个药物对癌细胞的影响)和你的数据集(比如,单细胞测序数据),剩下的事情,它自己搞定。
它的核心是一套多 Agent 系统,这很像我们之前讨论过的分子设计平台,但应用场景的复杂度提升了一个数量级。这个 AI 团队里有负责「分析任务」的,有负责「设计方法」的,还有负责「执行实验」的。最妙的是,它们之间不是简单地分工,而是一个「图谱式讨论」架构。这意味着「方法设计师」提出的一个模型方案,会立刻被其他 Agent 从不同角度进行批判和审视,然后进行修改和完善。这不就是我们项目组开会的日常吗?只不过效率高了无数倍,而且 24 小时不知疲倦。
这种协作式推理带来的结果是惊人的。在单细胞扰动预测这个药物研发的核心领域,CellForge 的表现堪称恐怖。预测基因敲除、药物处理或细胞因子刺激后的细胞转录组变化,错误率直接降低了 40%。这意味着我们能更准确地在早期阶段就判断一个药物靶点是否值得继续投入,或者一个候选药物会不会产生意想不到的脱靶效应。
对于研发人员来说,CellForge 这样的工具如果能成熟落地,其意义是颠覆性的。它将彻底改变计算生物学家的工作模式。我们不再需要把大量时间浪费在调参和写 Pytorch 代码上,而是可以回归科学的本源:提出更有洞察力的生物学问题,设计更巧妙的实验方案。AI 负责繁重的「工程」部分,人类则专注于「科学」部分。
当然,这篇论文刚挂在 arXiv 上,我们还得看看它在更多、更复杂的真实世界数据集上的表现。
📜Paper: https://arxiv.org/abs/2508.02276
2. AI 给蛋白画「藏宝图」,告诉我们药往哪儿放
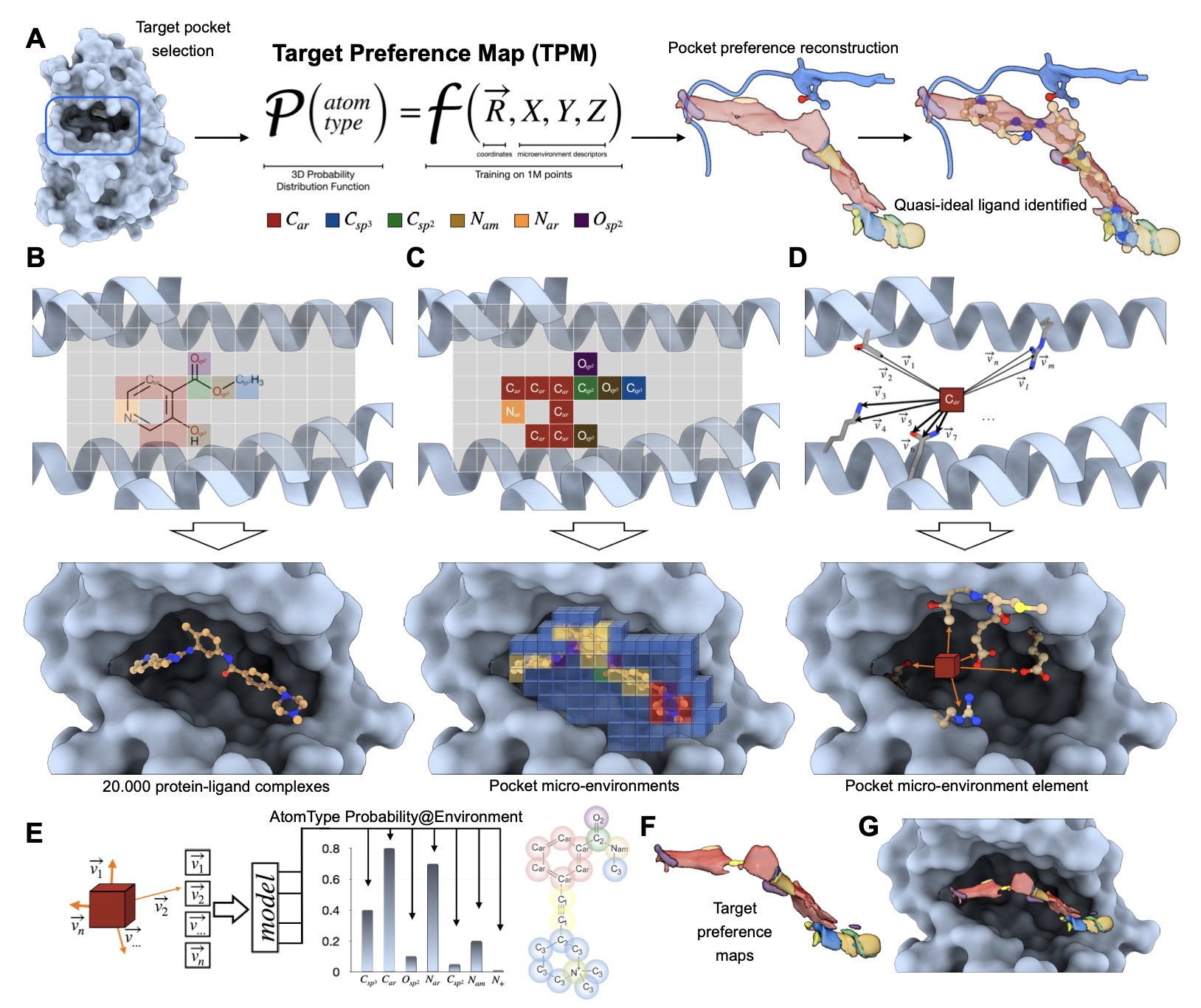
药物化学大部分时间都像是在玩一局三维版的「我画你猜」。我们盯着电脑屏幕上那个蛋白质的活性口袋——一堆乱七八糟的螺旋和折叠——然后开始凭着直觉和经验瞎蒙:「嗯……这里看起来像个疏水区,我应该塞一个苯环进去。哦,那边那个角落里有个谷氨酰胺,也许我可以在我的分子上加个能跟它形成氢键的基团?」这其中充满了「也许」、「可能」和「试试看」,更像是艺术创作,而不是严谨的工程。
好了,现在想象一下,有一个工具,它能拿过你手里的蛋白结构,然后给你生成一张高清的 3D「倾向图」。地图上清清楚楚地标着:这片红色区域,极度渴望一个氢键受体;那片蓝色区域,特别喜欢带正电荷的基团;而这块绿色的地方,你最好放点疏水性的东西,别的免谈。这张图,就是这篇论文里提到的「靶点偏好图」(Target Preference Maps, TPMs)。它把蛋白质口袋那晦涩的结构语言,翻译成了化学家能听懂的、关于「化学偏好」的直白描述。
它是怎么做到的?
它没有去学习完整的药物分子和蛋白质的结合模式。那种方法充满了历史偏见——因为化学家总是倾向于做一些熟悉且容易合成的骨架,AI 学到的可能只是「人类化学家的合成偏好」,而不是真正的物理化学原理。
所以,作者们换了个思路。他们把超过一百万个已知的配体 - 蛋白相互作用,打碎成了无数个微小的「原子微环境」。模型学习的不是「这个叫伊马替尼的分子是如何结合的」,而是更本质的东西,比如「在一个疏水环境里,被两个芳香环夹着的羰基氧原子,它周围最喜欢出现什么样的原子?」通过学习这些底层的、普适的物理规则,这个模型获得了惊人的泛化能力。
当然,只说不练假把式。任何一个计算模型,如果不能在现实世界里证明自己,那它就只是一堆漂亮但无用的代码。这篇论文做了前瞻性验证。
他们挑了一个硬骨头:PEX14-PEX5 蛋白 - 蛋白相互作用界面。这种靶点表面又大又平,没啥特征,是药物化学家的噩梦。他们用 TPMs 指导设计了新一代的抑制剂,然后走进实验室,把它们合成出来,拿去测活性。
结果呢?新分子的活性确实得到了显著提升。
这个模型目前有个短板:它对化学键「一无所知」。它能告诉你「在这里放个碳,旁边放个氮」,但它不知道你是否能用现实中的化学反应,稳定地把这两个原子连在一起。这就像一个建筑 AI,设计了一座无比酷炫的悬浮城堡,却完全没考虑过重力和材料强度。最后,还是得满身化学试剂味道的「包工头」,去想办法把图纸变成现实。
而且,它依赖的是静态的晶体结构,可我们都知道,蛋白质在体液里可不是一动不动的雕塑,它们在不停地摆动、呼吸。
📜Title: A Universal Model for Drug-Receptor Interactions
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.01.668090v1
3. AIDD 新思路:别再假装分子活在真空中
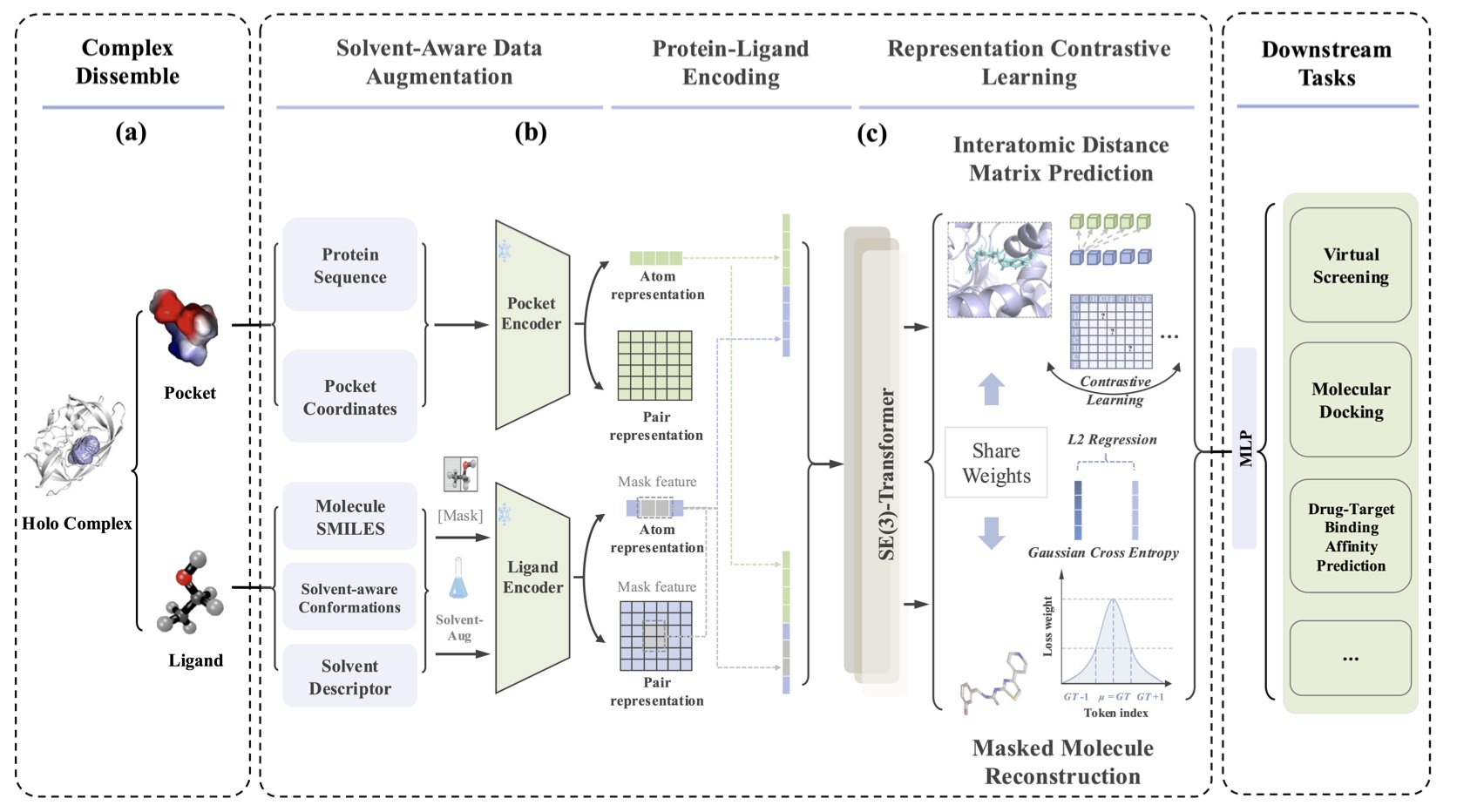
几十年来,我们的很多计算模型都有一个原罪:它们假装分子活在一个完美的、空无一物的真空中。我们画出漂亮的结合口袋,把小分子塞进去,计算各种静电力和范德华力,然后得出一个结合能。但我们都心知肚明,在真实的细胞中,这一切都发生在一锅由水分子主导的、拥挤不堪的「汤」之中。
水分子无处不在,它们会拉扯、推挤、包裹你的药物分子,彻底改变它的「心情」和「构象」。一个在真空中看起来完美的氢键,在水溶液里可能毫无优势,因为水自己就能形成更好的氢键。这种「脱溶剂化」的代价,是我们在药物设计中永远无法回避的幽灵。
这篇论文的核心武器叫「溶剂感知的数据增强」。
名字听起来挺唬人,但想法很直观。它不再只给 AI 看一张分子的「标准照」,而是把它扔进各种不同的「溶剂浴」里,让它充分折腾,然后拍下一系列它在这些环境里可能摆出的各种构象。通过这种「特训」,AI 学会了识别一个分子的本质,而不是它在某种理想状态下的单一形象。它终于开始理解,一个分子的柔性以及它与环境的互动,是它身份的一部分。
这个框架还采用了对比学习和多任务学习。这意味着它不仅学习识别分子,还要完成分子重建、原子间距离预测等多个任务。这就像训练一个侦探,不只让他认人,还要让他能根据零碎线索画出肖像、推断身高。这种全方位的训练,让模型对分子的理解更深刻。
数据确实漂亮。在 PoseBusters 这个堪称对接领域「驾考科目二」的基准上,成功率达到 82%。在虚拟筛选任务中,AUC 达到 97.1%。还有一个案例研究,对接的 RMSD 误差小到 0.157 埃,这已经进入了亚埃级别,足以让任何一个计算化学家心跳加速。
当然,这些都是在标准数据集上跑出来的「闭卷考试」成绩。真正的考验,是把它扔到我们自己正在进行的新药项目中,面对那些全新的、充满未知的靶点和分子,看它是否还能如此神勇。
💻Code: https://github.com/1anj/SolvCLIP
📜Paper: https://arxiv.org/abs/2508.01799