
目录
- DREAM-GNN 通过一个双通道架构,同时分析药物 - 疾病的已知关系网和它们各自的内在特征,极大地提升了老药新用预测的准确性和可靠性。
- DecoyDB 创造了一个包含数百万个「错误答案」的庞大数据集,让 AI 通过对比学习,真正理解了分子结合的「正确姿势」,极大地提升了亲和力预测的准确性。
- 计算和 AI 正在为那些曾被判死刑的「不可成药」靶点带来新武器,从变构调节到蛋白降解,游戏规则已经改变。
1. DREAM-GNN:老药新用的双通道高速公路
老药新用,这个概念听起来就像是炼金术士的梦想:用已经证明安全的旧药,去治疗一个全新的疾病。省钱,省时,简直完美。
但实际操作起来,这活儿跟大海捞针差不多。计算方法喊了很多年,但很多模型要么太天真,要么太偏科。
有的模型只看关系网,比如 A 药治 X 病,B 药治 Y 病,那么和 A 药结构相似的 C 药是不是也能治 X 病?有的模型只看特征,比如 D 药和 E 药都能抑制某个激酶,那么它们可能治疗同一种癌症。这两种思路都有道理,但都只看到了一半的真相。
DREAM-GNN 的方法很简单直接:小孩子才做选择,成年人全都要。
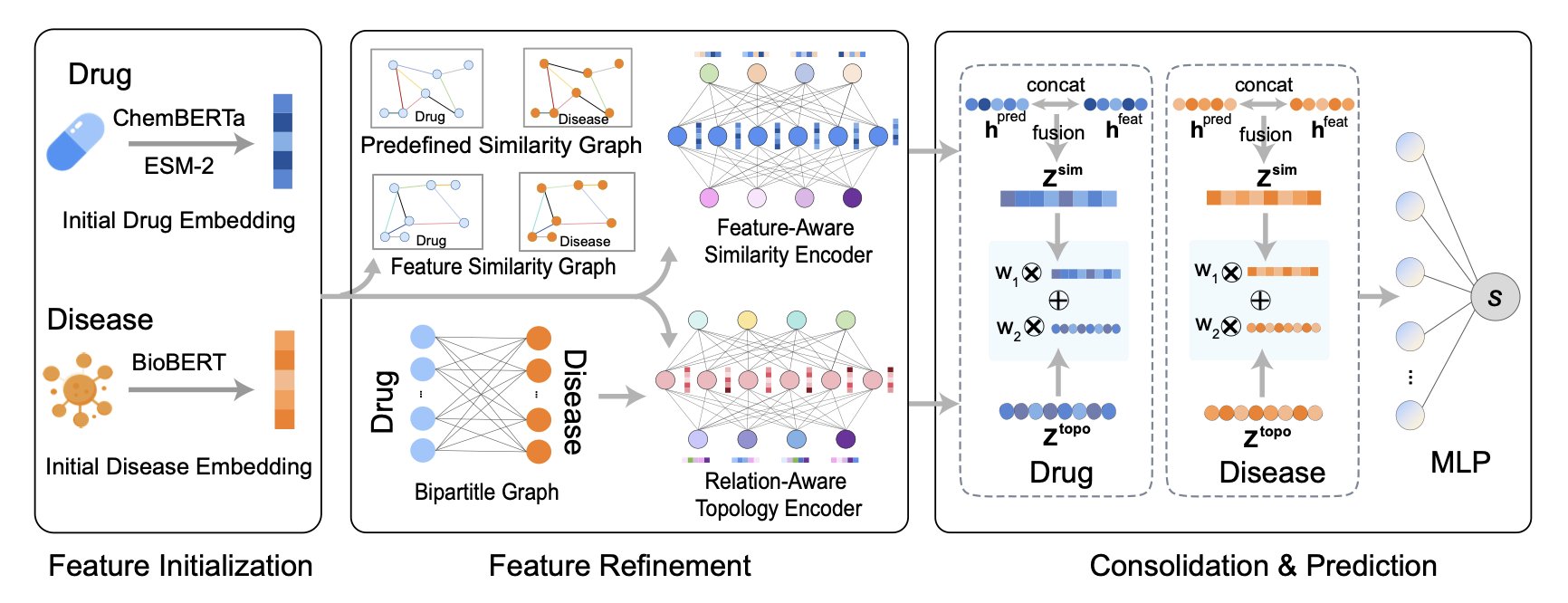
它设计了一个「双通道」架构,这简直是神来之笔。
第一条通道,是个关系专家。它构建一个巨大的图谱,上面是所有已知的药物 - 疾病关联,它专门学习这个网络拓扑结构里的门道,像个社交网络分析师一样,研究「谁和谁有关系」。
第二条通道,是个特征专家。它不看已知的治疗关系,而是深入研究药物和疾病的内在属性。它怎么研究?它不是自己从头瞎琢磨,而是请来了一帮「外援」:用 ChemBERTa 来读懂小分子的化学语言,用 ESM-2 来理解靶点蛋白的生物学功能,用 BioBERT 来解析疾病的医学文本描述。这些都是在各自领域身经百战的预训练模型。DREAM-GNN 把这些专家的见解整合起来,去判断哪些药物「长得像」,哪些疾病「病理相似」。
最后,这两条高速公路上跑出来的信息,在一个交叉路口汇合。模型综合了「关系网」和「相似性」两方面的情报,给出的预测自然就比那些「独眼龙」模型要靠谱得多。
数据也证明了这一点。在多个标准数据集上,DREAM-GNN 的性能都名列前茅。更重要的是,它在 AUPRC 这个指标上表现突出。这个指标对于搞实际应用的人来说至关重要,因为它能真实反映模型在数据极度不平衡(绝大多数药物和疾病没关系)的情况下,找到那几个真正「金子般」的阳性配对的能力。
📜Paper: https://www.biorxiv.org/content/10.1101/2025.07.07.663530v1
2. DecoyDB:给 AI 一本高质量「错题集」
在药物发现的计算世界里,预测一个小分子和蛋白的结合亲和力,是我们永恒的痛。
我们有各种花哨的图神经网络(GNN)模型,但它们大多都营养不良——因为高质量的、带标记的训练数据实在是太少了。这就像你想训练一个顶级的艺术品鉴赏家,却只给他看了三幅画。
对比学习(Contrastive Learning)是一个很时髦的解决方案。它不用告诉模型「正确答案」是什么,而是给它看一对东西,让它判断「它俩像不像」。对于分子结合,就是让模型学会区分「好的结合模式」和「差的结合模式」。
问题来了,我们有「好的」(晶体结构),但从哪找那么多「差的」来给它作对比呢?
过去的做法很粗暴:把正确的结构拿过来,随机晃一晃、扭一扭,造一个「假的」出来。
这方法的问题是,你造出来的很多构象在生物化学上根本就是胡闹,能量高得离谱。用这种「垃圾」负样本训练模型,就像教一个孩子识别猫,是通过给他看一张猫和一张被打碎的马赛克图片。孩子最后学会的不是识别猫,而是识别马赛克。
DecoyDB 尝试解决这个问题。
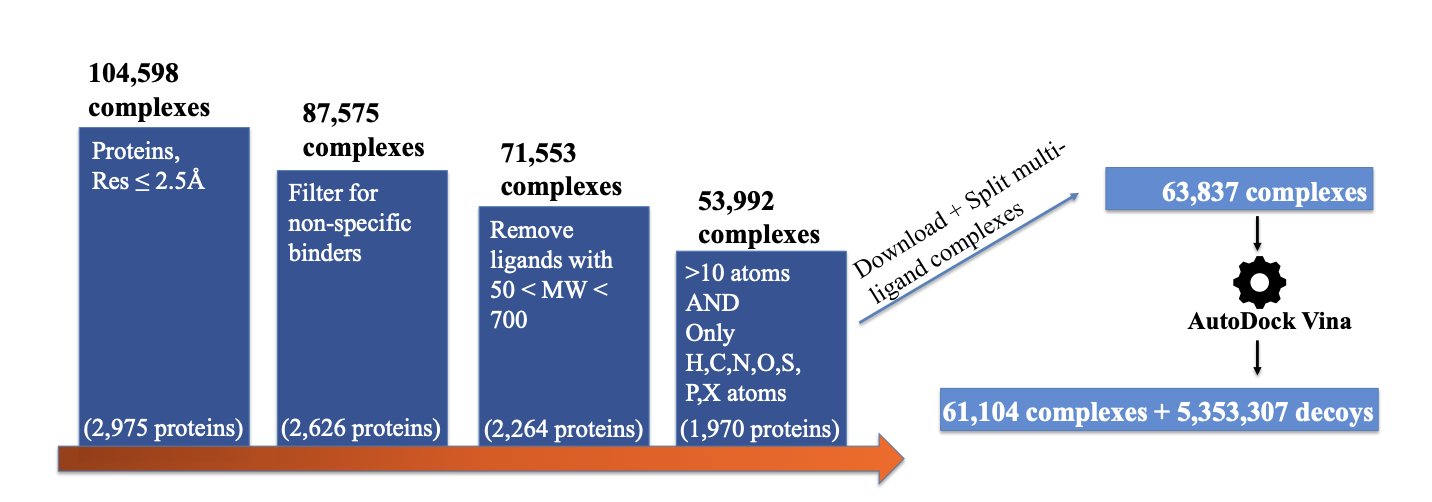
他们用计算方法,生成了超过 500 万个「诱饵」(Decoy)结构。这些诱饵,都是看起来「似乎有道理」,但实际上是次优的结合模式。就像教孩子识别猫时,我们不给他看马赛克,而是给他看狗、老虎和狐狸。这些都是更高级、更有意义的「错误答案」。
他们还给每一个「错误答案」都打上了一个标签:RMSD,也就是它和标准答案(真实结构)的偏差程度。这就不仅仅是告诉模型「这是错的」,而是告诉它「这个错得比较离谱,那个错得只有一点点」。这一下子就把训练信息的含金量,提升了好几个数量级。一个粗糙的二元判断题,变成了一道信息量丰富的问答题。
光有好的「教材」还不够,还得有好的「教法」。研究者们配套开发了一个新的对比学习框架。它有一个「双轨制」的损失函数,既能让模型在「对」和「错」之间做宏观判断,也能在「错」和「更错」之间做细微辨析。更妙的是,他们还加入了一个基于物理的约束(去噪分数匹配),这等于在旁边站了个物理老师,不断提醒模型:「你给出的答案,不仅要长得好看,还得符合能量最低原理,得稳定才行!」
结果怎么样?
用 DecoyDB 预训练过的模型,在亲和力预测任务上,无论从准确性、泛化能力还是数据效率上,都把之前的模型打得落花流水。
DecoyDB 为整个领域提供了一套宝贵的基础设施,一个高质量的、开放的「错题集」。有了它,我们终于可以训练出真正「懂行」的 AI 鉴赏家,而不只是一个「找茬」专家。
📜Title: DecoyDB: A Dataset for Graph Contrastive Learning in Protein-Ligand Binding Affinity Prediction
📜Paper: https://arxiv.org/abs/2507.06366
💻Code: https://github.com/spatialdatasciencegroup/DecoyDB
3. AI 正在抹除「不可成药」这个词
「不可成药」(undruggable)这个词就像是一纸判决书。一旦一个和疾病高度相关的蛋白靶点被贴上这个标签,基本上就意味着它会被打入冷宫,成为无数项目坟场里的一块墓碑。
为什么?
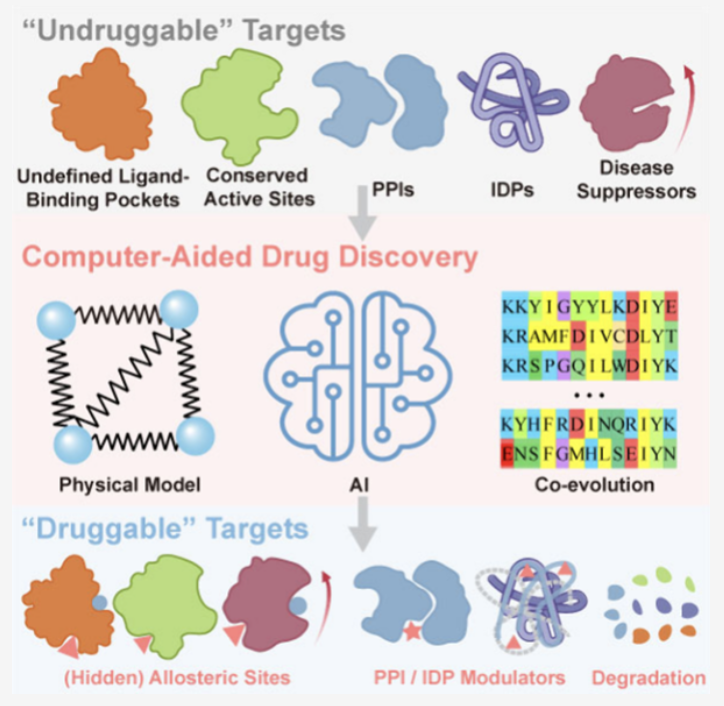
因为它们要么没有一个可供小分子「安家」的漂亮口袋,要么像一碗煮烂的意大利面条一样毫无固定形状(本质上是本质无序蛋白,IDP),要么就是和其他蛋白的结合面又大又平,像一堵墙,让小分子无处下手(蛋白 - 蛋白相互作用,PPI)。
几十年来,我们只能眼巴巴地看着这些靶点——比如 c-Myc——在疾病中兴风作浪,却束手无策。
但这篇综述告诉我们,时代变了。计算生物学和 AI,正在把这份判决书一张一张地撕掉。
首先,我们改变了攻击的思路。
对于那些像面条一样的IDP,过去的我们认为没法搞,因为它没有稳定的「锁孔」。现在的计算方法,比如基于系综的筛选,不再试图去捕捉一个静止的构象,而是去理解它整个「舞蹈」的过程,在它动态变化中找到可以下手的脆弱瞬间。c-Myc 的抑制剂(比如 Omomyc)能进入临床,就是最好的证明。
对于那些像墙一样的PPI界面,我们不再幻想用一个「小石子」去堵住一整面墙。计算工具能帮我们在这面墙上进行「地质勘探」,找到那些微小的、通常被忽略的「裂缝」或「热点」,然后设计出能精准楔入其中的分子。
其次,我们开发了全新的武器。
如果正门(活性位点)堵不上,我们就找后门。变构调节就是这个思路。计算模拟可以帮助我们预测蛋白上那些远离活性位点,但又能远程遥控其功能的「电路开关」。找到它,然后用一个小分子去拨动它,就能四两拨千斤。
如果连后门都找不到呢?那就用最狠的一招:靶向蛋白降解(TPD)。PROTACs 和分子胶水就是这类武器的代表。我们不再想着去「抑制」这个坏蛋白,而是直接把它「干掉」。我们设计一个双头分子,一头抓住靶蛋白,另一头抓住细胞里负责清理垃圾的 E3 连接酶,像一个「分子手铐」把两者铐在一起,然后让细胞自己的「垃圾焚烧炉」把它彻底清除。而设计这个「分子手铐」的长度、角度和化学性质,计算模拟在其中扮演了至关重要的角色。
这篇综述清晰地表明,我们正处在一个范式转移的关口。「不可成药」这个词,正在慢慢变成一个历史术语。它反映的不是靶点本身的属性,而是我们过去工具箱的局限性。现在,有了计算和 AI 这些新工具,那份长长的「不可成药」清单,正在变成一份充满机遇的「尚待成药」(yet-to-be-drugged)的项目清单。
📜Paper: https://pubs.acs.org/doi/10.1021/acs.chemrev.4c00969