
目录
- 这个开源工具包使用简单的 YAML 配置文件,让搭建复杂、可复现的多模态生物学基础模型,从一项工程挑战,变成像搭乐高积木一样的探索。
- ChatMDV 将研究者的自然语言直接翻译成可执行代码,让复杂的生物信息学数据可视化,从少数专家的技能,变成每个实验室成员都能上手的工具。
- DiffGui 在生成原子的同时构建化学键,并以结合力和类药性作为引导,使 AI 生成的分子成为化学结构可信的实体,而非简单的原子云。
- 一个 AI 框架通过「达尔文式」进化循环优化分子,同时通过「哥德尔式」元学习循环优化自身的药物发现流程。
- 通用大模型能解决不少标准生信问题,但它们更像博览群书的学生,而非能独立思考的研究员。
1. AIDO.ModelGenerator:生物多模态模型开发的乐高
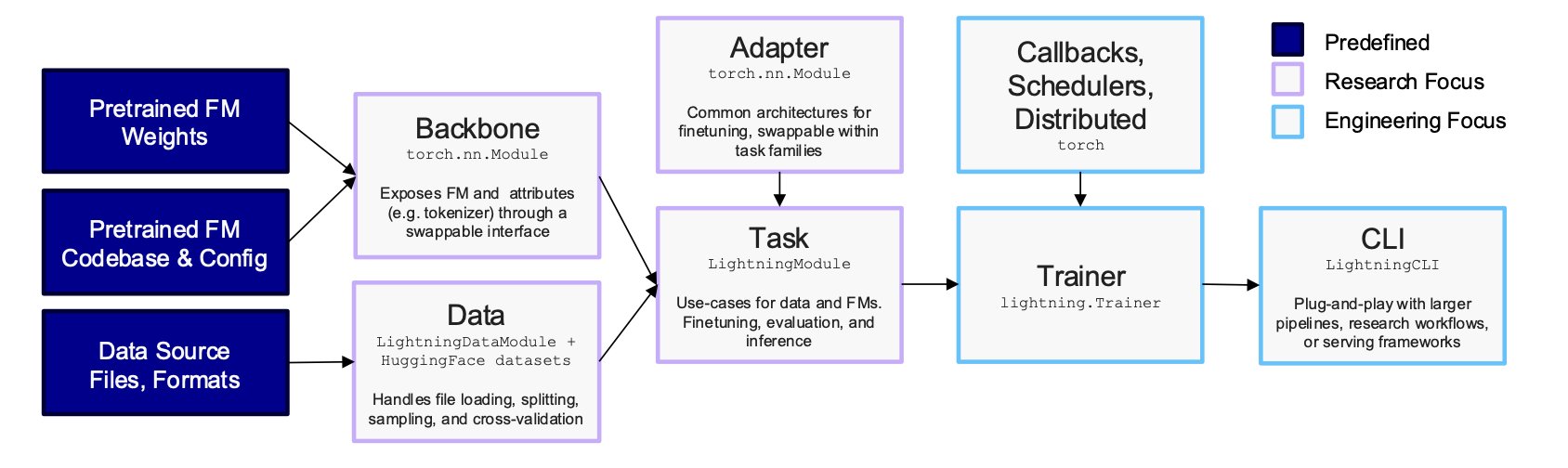
生物研究者常常被计算和工程问题所困。尤其在大模型时代,搭建一个融合 DNA、RNA 和蛋白质等多种数据的模型,过程复杂。研究者需要身兼机器学习工程师与软件运维专家,才能开始真正的生物学工作。整个过程繁琐,结果难以复现,拖慢了科学发现的进程。
AIDO.ModelGenerator 这款新工具直面这个痛点。它的核心思路是将复杂流程标准化、模块化,如同为生物基础模型开发的乐高积木。用户无需从零开始编写模型搭建、融合与训练的代码,只需准备一个 YAML 配置文件。
例如,要用一个 30 亿参数的 DNA 模型和一个 5 亿参数的 RNA 模型进行交叉注意力融合,只需在 YAML 文件中声明。想在单张 A100 显卡上,通过参数高效微调(PEFT, Parameter-Efficient Fine-Tuning)技术运行大模型,也只需几行配置。这把技术门槛从机器学习专家,降低到能编写配置文件的科学家。
在克罗恩病(Crohn’s disease)的案例中,该工具展现了它的能力。传统的差异表达分析,在近 19000 个基因中,将已知的临床靶点 SOX4 排在第 6068 位,如同大海捞针。AIDO.ModelGenerator 通过模拟基因敲除(in-silico knockout),将 SOX4 的排位提升至第 14 位。这种提升是质的飞跃,为寻找靶点提供了明确的线索。
在 RNA 剪接预测任务中,该工具同样表现出色。仅使用单一模态数据,好比依据静态地图预测实时交通。通过融合 DNA(基因组静态蓝图)与 RNA(动态表达信使)数据,模型的预测性能提升超过 10%,达到了新的业界最佳水平(SOTA, State-of-the-Art)。这种多模态方法为理解复杂生物系统提供了更全面的视角。
该工具对可复现性的重视解决了计算生物学的一个核心痛点。学术研究中,实验结果难以复现是一个普遍问题。AIDO.ModelGenerator 通过锁定的配置文件和确定性运行,保证了每次实验都能获得字节级别完全相同的结果。这使得研究发现可靠、可验证,也方便同行与审稿人重现整个工作流程,体现了科学研究的严谨性。
AIDO.ModelGenerator 本质上是一个加速器和工具集,它将生物学家从繁重的工程任务中解放出来。研究者可以因此专注于生物学问题本身——提出假说、设计实验、验证发现,无需在调试代码和配置环境上耗费过多时间。
📜Title: Rapid and Reproducible Multimodal Biological Foundation Model Development with AIDO.ModelGenerator
📜Paper: https://www.biorxiv.org/content/10.1101/2025.06.30.662437v1
💻Code: https://github.com/genbio-ai/ModelGenerator
2. ChatMDV:让生物信息学分析告别代码
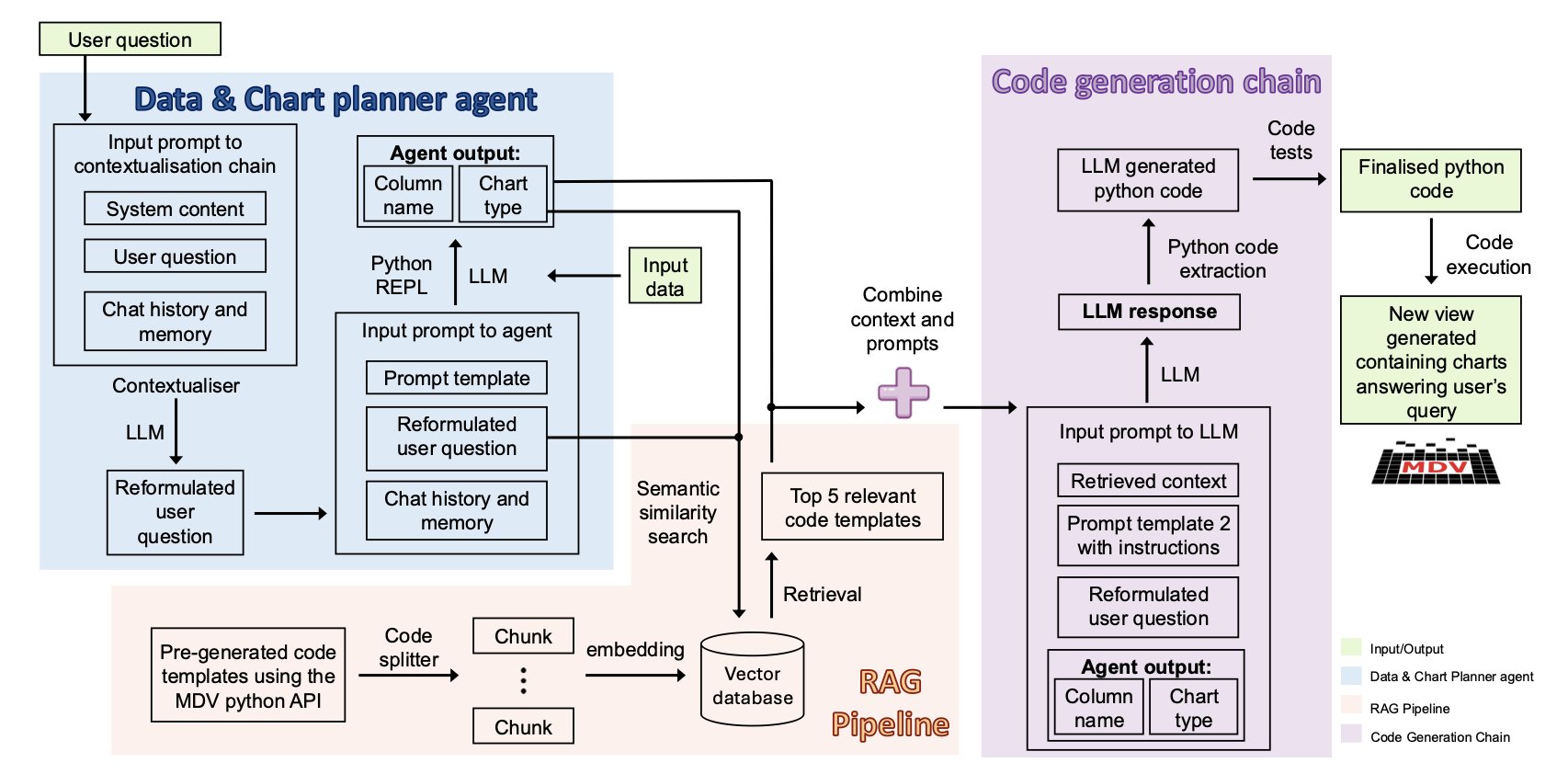
生物信息学分析的门槛很高,要求研究者既懂生物又会编程,这拖慢了科研进度。ChatMDV 提供了一个解决方案。它如同一个翻译官,使用大语言模型(Large Language Model, LLM)和检索增强生成(Retrieval-Augmented Generation, RAG)技术,将一句「用 UMAP 图展示这些细胞簇的 A 基因表达情况」这样的自然语言,直接翻译成生成图表的 Python 代码。数据分析由此变成一场对话,湿实验科学家可以亲自、快速地探索数据、验证想法,无需再排队等待生物信息学家的协助。
生物学实验室里存在一道无形的鸿沟。一边是辛苦获得的单细胞测序数据,另一边是揭示生物学规律的图表和统计结果。横亘其间的是一堵由 Python、R 和复杂软件包砌成的高墙。
研究者想跨越这堵墙,要么花几年时间学习编程,要么将宝贵的数据交给那位日程永远排满的生物信息学家,然后开始等待。
ChatMDV 的目标,就是拆掉这堵墙。
AI 当翻译,你只管提问
ChatMDV 的思路很直接:让 AI 学习生物学家的语言。它把自己定位成一个顶级的同声传译。
它的工作流程如下:
研究者用简单的自然语言提出需求,例如,「用 UMAP 图显示这些细胞,并根据细胞类型给它们上色」。
ChatMDV 内部的「规划智能体」会将这句话拆解成一个清晰的行动计划:「用户需要一张 UMAP 图,按‘cell_type’列的数据进行着色。」
接着,一个「代码生成」模块开始工作。它并非凭空编写,而是通过检索增强生成(RAG)流程,在预设的「代码库」和「知识库」中查找最相关的代码片段和函数用法。这好比一个开卷考试的学生,他不需要记住所有知识点,只需知道去哪里查阅,然后正确地组合信息。
最后,它生成一段 Python 代码并自动执行,将你想要的 UMAP 图呈现在眼前。
不只是聊天,还能动手
ChatMDV 生成的图表呈现在一个交互式查看器(MDV)中。AI 完成初步绘图后,你还可以像使用普通软件一样,用鼠标点击、缩放、筛选,对图表进行微调。
这种「自然语言输入 + 图形界面微调」的组合,降低了使用门槛,也预示了未来科学软件的形态。
到底靠不靠谱?
研究团队用三个复杂度递增的真实数据集对 ChatMDV 进行了测试,从简单的 PBMC 数据到复杂的肺癌图谱,它都表现出很高的成功率。在最简单的任务上,成功率达到 100%。
这表明,它有能力处理真实科研中那些不完美的数据。
虽然 AI 还无法完全理解所有模糊的科研设想,但 ChatMDV 证明,科学家与数据之间无代码的对话式交互是可行的。
它会把生物信息学家从大量重复性的初级可视化任务中解放出来,使其能够专注于解决更需要智慧与创造力的科学问题。
📜Title: ChatMDV: Democratising Bioinformatics Analysis Using Large Language Models
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.26.671083v1
3. DiffGui:AI 造分子,先画骨再填肉
与 3D 分子生成模型打过交道的人都了解一个现实:这些 AI 擅长排列原子,却不精通化学。
给定一个蛋白质口袋,AI 能生成一团在三维空间中填充良好的原子云。但将这些原子云转换成带化学键的二维结构图时,问题便出现了:五配位碳、不存在的化学键等违反化学常识的结构屡见不鲜。这如同建筑师只标出柱子位置,却忽略了梁与楼板,使建筑无法成立。
Nature Communications 的一篇论文介绍的方法,旨在让 AI 在放置原子的同时,也构建起化学键的框架。
先画骨,再填肉
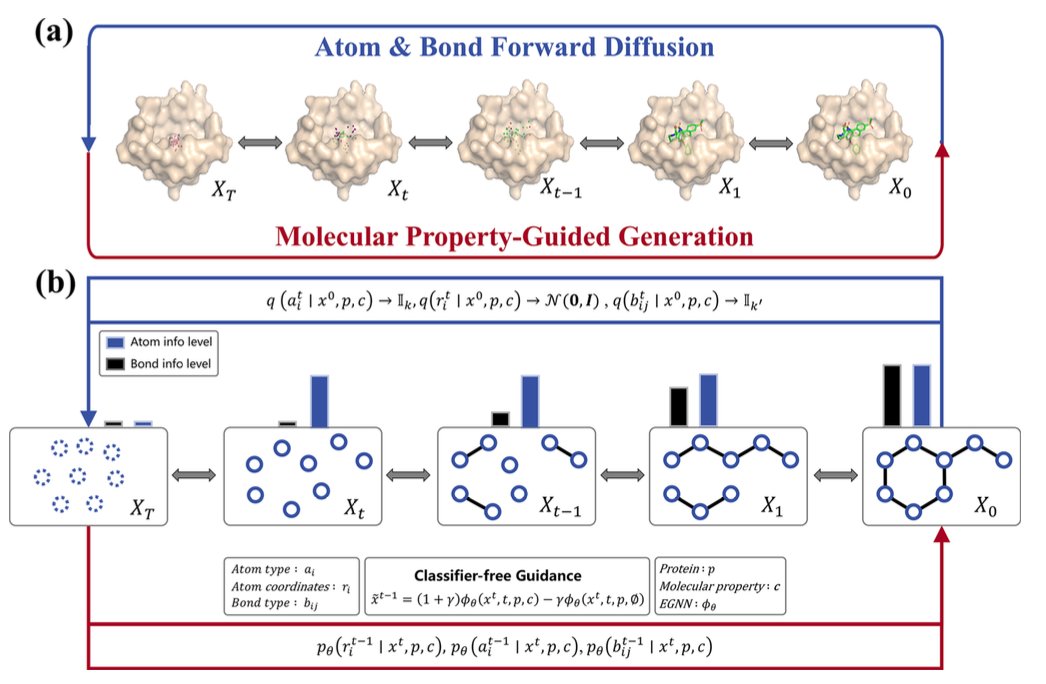
新模型名为 DiffGui,其核心是同步生成原子和化学键。
它属于扩散模型。传统扩散模型从一团模糊的原子「像素点」开始,逐步使其清晰。DiffGui 则从模糊的「像素点」与模糊的「连接线」同时开始。
在生成的每一步,模型同时确定原子的类型和位置,以及原子间的化学键。
这样,化学键成为生成过程的内在约束,而非后续添加的步骤,从根本上保证了最终分子的化学结构合理性。
给 AI 一个「指南针」
仅有化学上合理的骨架并不足够,目标是生成一个好的分子。
DiffGui 引入了属性引导 (property guidance) 机制。在分子生成的每一步,多个属性评估器会提供反馈。
例如,「结合亲和力」评估器判断当前步骤是否增强了分子与靶点的结合,并引导其朝结合更紧密的方向优化。「类药性」评估器则会审视分子的化学性质,如溶解度或氢键特征,并进行相应调整。
通过这种持续的多维度引导,生成过程从单纯模仿训练数据,转变为一个有目的、主动朝向「好分子」的优化过程。
这东西真的靠谱吗?
同时构建骨架并使用指南针引导,使 DiffGui 生成的分子质量得到提升。
在一系列基准测试中,DiffGui 的表现超过了现有方法。
在一个真实的药物设计案例中,研究者使用该工具,为一个发生突变的蛋白质口袋成功设计出能够适应性结合的新分子。这表明 DiffGui 能够理解并响应精细的化学环境变化,是一个实用的设计工具。
📜Title: Target-aware 3D Molecular Generation Based on Guided Equivariant Diffusion
📜Paper: https://www.nature.com/articles/s41467-025-63245-0
💻Code: https://github.com/QiaoyuHu89/DiffGui
4. AI 药物发现:一个能自我进化的机器
AI 药物发现工具能生成分子、预测性质,但它们大多是静态的。模型一旦建成,性能就固定了,改进需要人工干预并重新训练。如同有一辆好车,想提速,还得自己动手改装引擎。
一个名为达尔文 - 哥德尔药物发现机 (Darwin–Gödel Drug Discovery Machine, DGDM) 的新框架,目标是造出一辆能自己改装引擎的车。
两个循环,两种进化
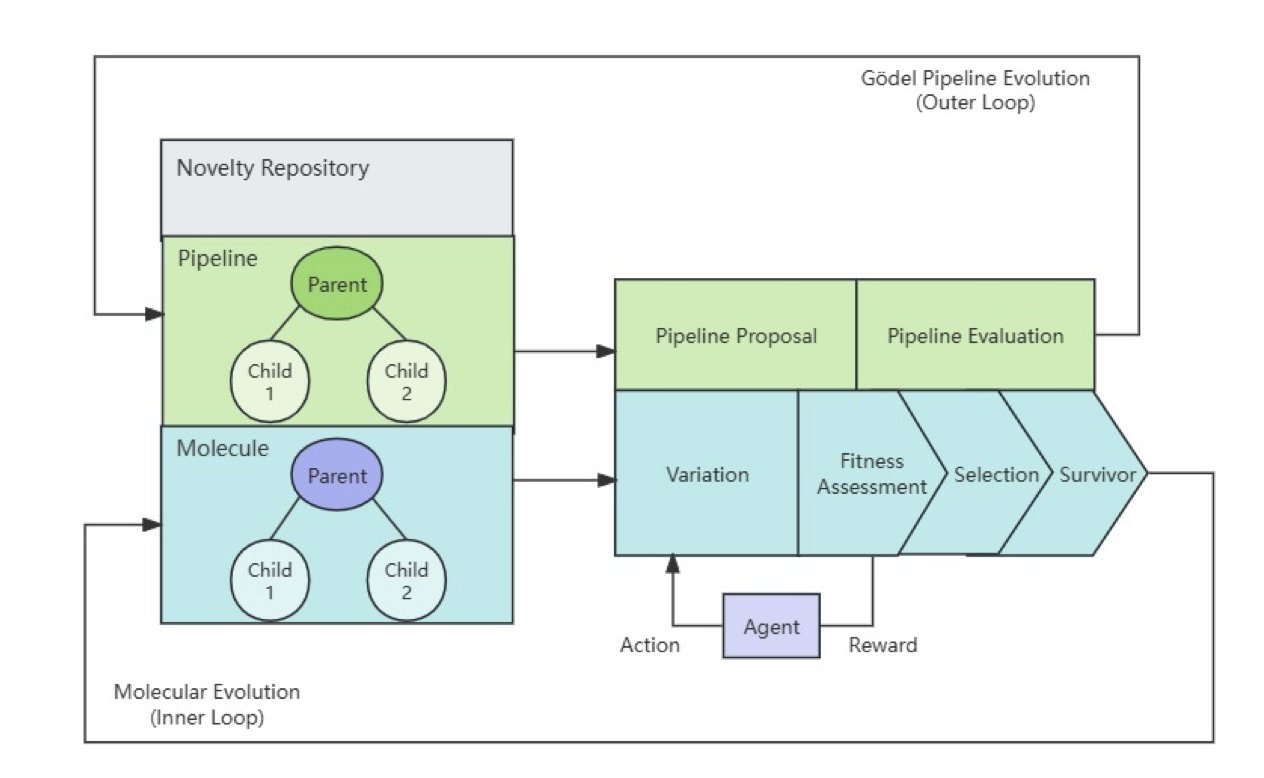
该框架的核心是两个相互嵌套的循环。
内循环:达尔文的世界
内循环优化分子,其工作方式如同微缩的达尔文进化。
首先,生成模型创造一批多样的候选分子。这些分子随即进入模拟的「自然环境」接受筛选,包括分子对接、结合亲和力预测和 ADMET 性质分析等计算评估。表现最佳的分子被选为「亲代」,用于生成下一代。
循环往复,分子在一代代进化中,愈发符合预设目标,例如结合力更强。
外循环:哥德尔的幽灵
外循环优化的不是分子,而是发现流程本身。
其灵感源于数学家哥德尔构想的「哥德尔机」——一种理论上能检查并修改自身代码、从而实现自我完善的机器。
现实中,无法用严格的数学逻辑证明流程更优,因此该框架采用统计学方法。外循环周期性地对整个药物发现流程提出修改建议,例如更换对接打分函数或调整分子生成策略。系统会通过统计检验,评估这些修改是否能提升整体性能。只有数据证明修改有效时,才会被采纳。这是一种由数据驱动、带风险控制的自我进化。
结果怎么样?
研究人员用该系统进行了一次概念验证。
经过双循环系统优化后,候选分子的中位结合亲和力获得提升,同时分子的类药性和新颖性保持在 100%。这表明系统在优化性能的同时,遵守了化学基本规则。
这仍是一项针对单个靶点的小规模概念验证。将湿实验数据整合进闭环,是下一步的挑战。
DGDM 展示了 AI 药物发现的一种可能方向:从使用 AI 工具,到构建能够自我完善的自主 AI 科学家。
📜Title: The Darwin–Gödel Drug Discovery Machine (DGDM): A Self-Improving AI Framework
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.21.671415v1
💻Code: https://github.com/deep-geo/DGDM
5. AI 参加生信考试,结果令人大跌眼镜
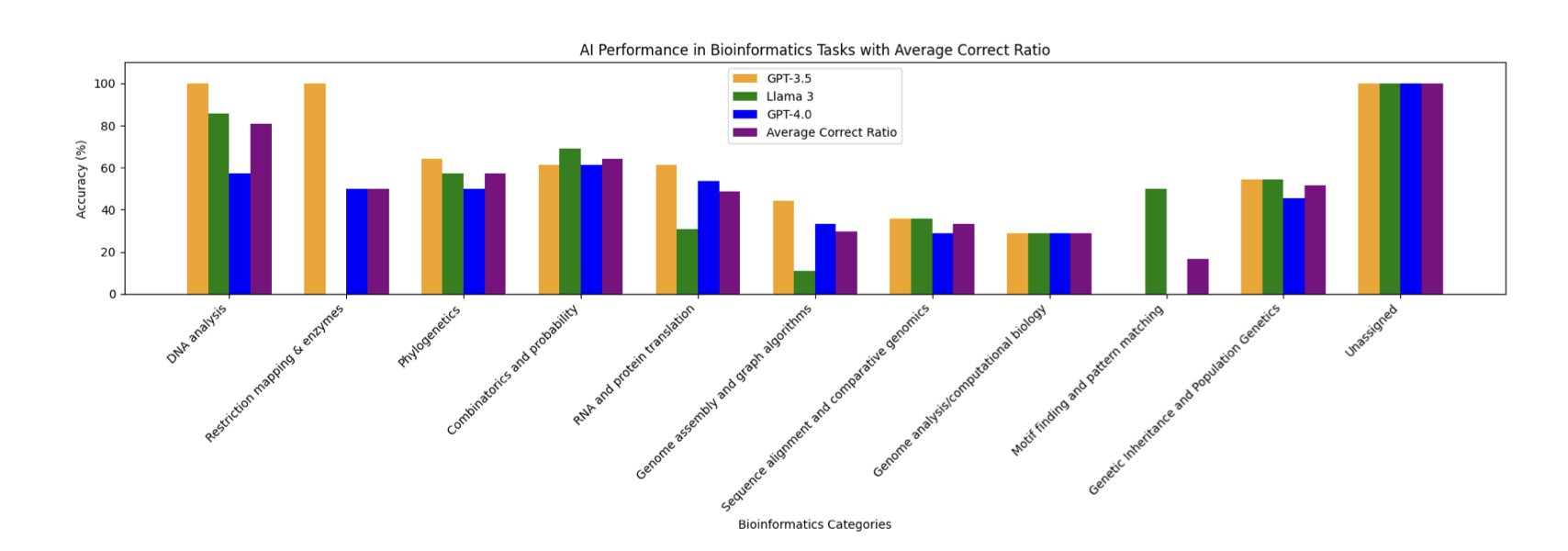
Rosalind 平台,是生物信息学学生的「奥数」训练场。它有上百个经典的计算问题,从计算 DNA 的 GC 含量,到寻找 RNA 的二级结构。学习生信的人,大多都在上面刷过题。
一篇新论文的研究者,将三个主流大语言模型 (Large Language Models, LLMs)——GPT-3.5、Llama-3-70B 和 GPT-4o——带到这个考场,测试了 104 道题目。
结果出人意料。
姜还是老的辣?
GPT-3.5拔得头筹,答对了 58% 的题目。而 GPT-4o 和 Llama-3 这两个更新、更强的模型,正确率反而只有 47%。
这并不意味着 GPT-3.5 比 GPT-4o 更聪明。一种可能的解释是,Rosalind 的题目是有标准答案的「课本习题」,GPT-3.5 的训练数据可能恰好包含了更多现成解法。更新的模型面对这些问题,或许会过度思考,尝试用更通用却非最优的逻辑求解。
AI 擅长什么,不擅长什么?
这次考试画出了当前通用大模型的「能力圈」。
在规则明确、计算直接的任务上,它们表现很好,例如计算 DNA 的各种统计特性。这好比让学生去做套公式的数学题,只要背过公式,基本不会出错。
但涉及更高阶推理的开放问题,比如基因组组装和序列比对,AI 就集体不及格了。这就像让学生去做需要多种思路和创造力的证明题,他脑子里的孤立公式,就不知道该怎么组合了。
这篇论文有一个发现很能说明问题:AI 解决一个问题的表现,与人类在 Rosalind 平台上尝试该问题的次数高度相关。
这就像一个学生,他在模拟考里做得最好的题,恰好都是他过去在各种辅导书上见过无数遍的原题。
这揭示了这些 AI 的学习方式:它们更接近于在庞大的记忆库中进行高效的模式匹配和信息检索,而非从第一性原理出发进行生物信息学推理。
所以,这些通用大模型仍然有用。对于学习基本概念的学生,它们是很好的助教。对于需要为标准化分析任务快速生成模板代码的研究员,它们是高效率的实习生。
📜Title: Out-of-the-box bioinformatics capabilities of large language models (LLMs)
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.22.671610v1