
目录
- UniDock-Pro 在一个统一的 GPU 加速平台上,首次整合了基于结构、基于配体和创新的混合虚拟筛选模式,实现了单 GPU 日筛百万分子的超高通量,同时显著提升了筛选的富集效率。
- XenoSignal 利用 AlphaFold3 实现了对异种移植模型中人 - 鼠细胞间相互作用的原子级精准预测,揭示了以往被忽视的物种特异性差异,这将深刻影响临床前研究的解读。
- TorchANI-Amber 通过一个巧妙的接口,让广泛使用的分子动力学软件 Amber,能无缝调用 ANI 系列神经网络力场,实现了在经典软件框架内进行「量子精度」的大规模生物分子模拟。
1. UniDock-Pro:GPU 加速的「三合一」虚拟筛选平台
药物发现的早期阶段,虚拟筛选(Virtual Screening)是最重要的「探路」工具之一。目标是从数百万甚至数十亿个分子的大型化合物库中,快速地「海选」出少数几个最有希望成为药物的候选分子。
传统的虚拟筛选,主要分为两大流派:
1. 基于结构的虚拟筛选(SBVS) :这就像是拿着一把「锁」(靶点蛋白结构),去试成千上万把「钥匙」(小分子),看哪个能插进去。经典工具如 AutoDock、Glide 就是干这个的。它很直观,但前提是你得有一把高质量的「锁」。
2. 基于配体的虚拟筛选(LBVS) :这更像是「照葫芦画瓢」。我们已经知道有几把「钥匙」能开这把锁,于是我们就去找和这几把「好钥匙」长得像的其他钥匙。经典工具如 ROCS 就是干这个的。它很快,但前提是你得先有几把「好钥匙」。
这两个流派,各有优劣,而且通常在不同的软件平台上实现。我们经常需要分别做一遍,再综合分析结果,流程繁琐。
UniDock-Pro 要打破这种壁垒,并把整个过程「开上高速」。
UniDock-Pro 的「三大法宝」
- 统一平台,GPU 加速 :它把 SBVS 和 LBVS 这两个核心功能,都整合到了一个统一的、基于 GPU 的计算框架里。通过最大化 GPU 的并行计算能力,它把筛选速度提升了几个数量级。在 LBVS 模式下,它的速度比 AutoDock-GPU 快了 43 倍。单卡一天处理上百万个分子,成为了现实。
- 更 LBVS :传统的 LBVS,在做形状匹配时,能量函数往往不够平滑,不利于高效的构象搜索。UniDock-Pro 实现了一种更平滑的、类似伦纳德 - 琼斯势的能量函数,让基于梯度的优化算法能跑得更顺畅。结果就是,在 DUDE-Z 这个「考场」上,它的早期富集率(EF1%)比传统工具高了 2.45 倍。
- 协同混合模式(Hybrid Mode) :这是 UniDock-Pro 最大的亮点。它不再让 SBVS 和 LBVS 各干各的,而是让它们「协同作战」。
协同混合模式是怎么运作的?
你可以把它想象成一个更高级的「寻宝」游戏。
UniDock-Pro 的混合模式,就是在分子对接的每一步,都同时参考这两张「藏宝图」。它会去寻找那些,既能很好地嵌入到「风水宝地」里,同时形状又和「金币」很像的分子。
这种方法,充分利用了两种信息的正交性和互补性,在几乎所有的基准测试中都取得了最高的富集效率。它能更有效地把真正的活性分子,从大量的「路人」分子中给挑出来。
UniDock-Pro 提供了一个「更准」、「更高效」的虚拟筛选新范式。对于需要在巨大的化学空间里快速淘金的药物研发人员来说,这是一个非常强大的武器。
📜Paper: https://doi.org/10.26434/chemrxiv-2025-bf5g7
2. XenoSignal: AlphaFold3 重塑异种移植模型药物研发
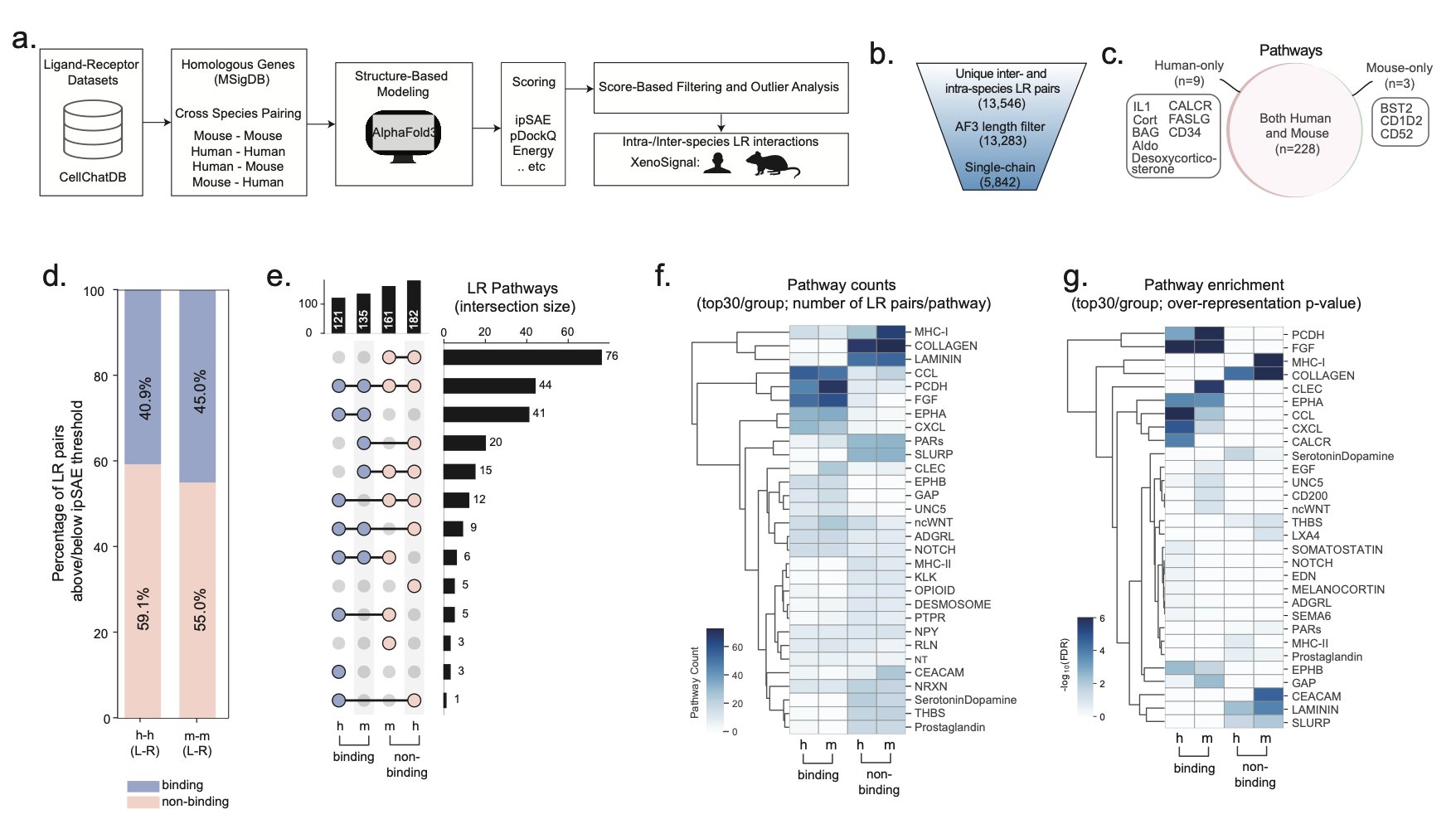
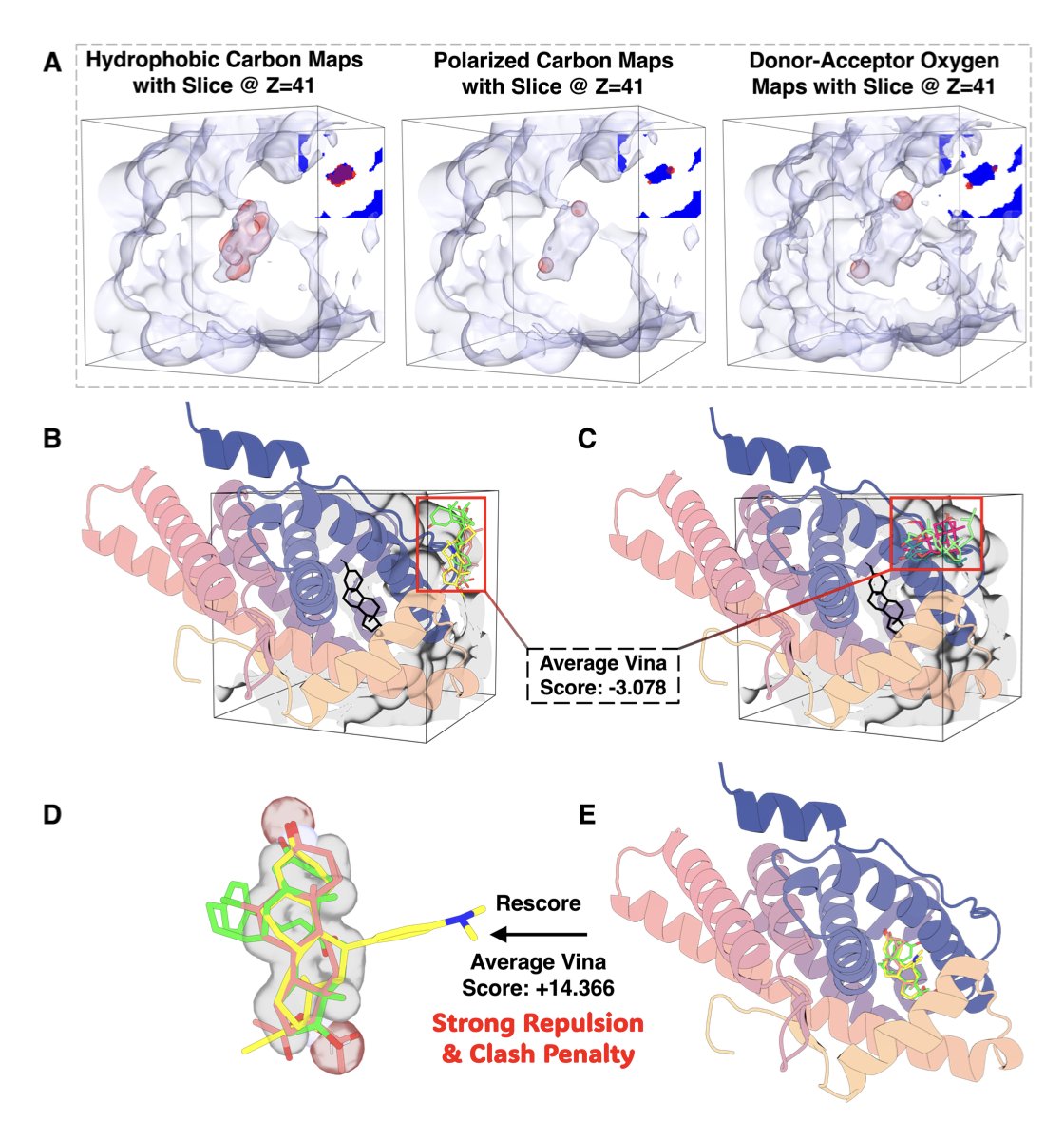
做临床前研究,尤其是肿瘤研究,谁都离不开异种移植模型(xenograft)。
我们把人的肿瘤细胞种到免疫缺陷的小鼠身上,然后用药,看看效果。这套流程用了几十年,但其实一直有个疙瘩:人的肿瘤细胞,真的能和周围的小鼠微环境「愉快地聊天」吗?我们一直默认,只要人和鼠的配体 - 受体蛋白序列足够像,它们就能相互识别和结合。但这个默认假设,就像是只看了两个人的简历就断定他们能合作无间,风险不小。
过去我们评估跨物种相互作用,多半是靠比对序列同源性。同源性超过 90%?那大概率能结合。这种方法简单粗暴,但忽略了一个根本问题:蛋白质的功能由三维结构决定,而不是一维序列。一个氨基酸的微小差异,就可能在关键的结合界面上造成一个巨大的空间位阻。
XenoSignal 用 AlphaFold3 把成千上万对人 - 鼠配体 - 受体复合物的原子级结构都给你算出来。这就相当于从看简历升级到了直接观察这两个人的互动录像。研究者发现,很多我们想当然认为可以跨物种结合的蛋白对,实际上结合得并不好,甚至完全不结合。
假设你开发了一个靶向人肿瘤微环境某条信号通路的药物,它通过阻断一个配体(来自肿瘤细胞)与一个受体(在小鼠基质细胞上)的结合来起作用。在小鼠模型里,你看到了漂亮的抑瘤数据。但 XenoSignal 的分析可能会告诉你,你药物靶向的这对人 - 鼠蛋白,其亲和力远低于人 - 人之间的亲和力。这意味着,你的药物在小鼠模型里可能只是「碰巧」有效,或者其真实的作用机制和你设想的完全不同。到了临床,面对纯粹的人体环境,药物的效果就可能大打折扣。
这个框架的厉害之处还不止于此。它还考虑了蛋白复合物的化学计量比(stoichiometry)和翻译后修饰这些更复杂的因素。比如,有些受体需要两个亚基才能工作,如果人和鼠的亚基无法正确组装,那后续的信号传递就无从谈起。
最终,XenoSignal 给出的不是一个模糊的「可能结合」的结论,而是一个经过物理模型严格计算的、高置信度的相互作用列表。这对于做药的人来说,价值巨大。我们可以在投入昂贵的动物实验之前,先通过计算筛选一遍,剔除那些最不靠谱的跨物种靶点,把资源集中在最有希望的方向上。
📜Title: XenoSignal: Investigating Intraand Inter-Species Ligand-Receptor Interactions Using AlphaFold3
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.13.670200v1
💻Code: https://github.com/MorrissyLab/XenoSignal
3. TorchANI-Amber:让经典模拟软件用上 AI 力场
做计算生物学和药物设计的领域,Amber 可以说是无人不知的「老牌贵族」。几十年来,无数重要的分子动力学(MD)模拟工作,都是在这个强大的软件平台上完成的。但 Amber 和很多经典 MD 软件一样,其核心依赖于经验性的经典力场。这些力场速度快,但在精度上,特别是处理一些精细的化学反应或复杂的相互作用时,总显得有些力不从心。
另一方面,以 ANI(ANAKIN-ME)为代表的新一代神经网络(NN)力场,正以其接近量子化学的精度,席卷着计算化学领域。但这些新兴的「AI 新贵」,往往活在独立的 Python 生态里,很难被集成到像 Amber 这样用 Fortran 和 C++ 构建的、庞大而复杂的系统中去。
这就造成了一个非常尴尬的局面:我们手里既有功能最全面、生态最成熟的模拟软件,又有精度最高的力场模型,但这两者却无法结合在一起。
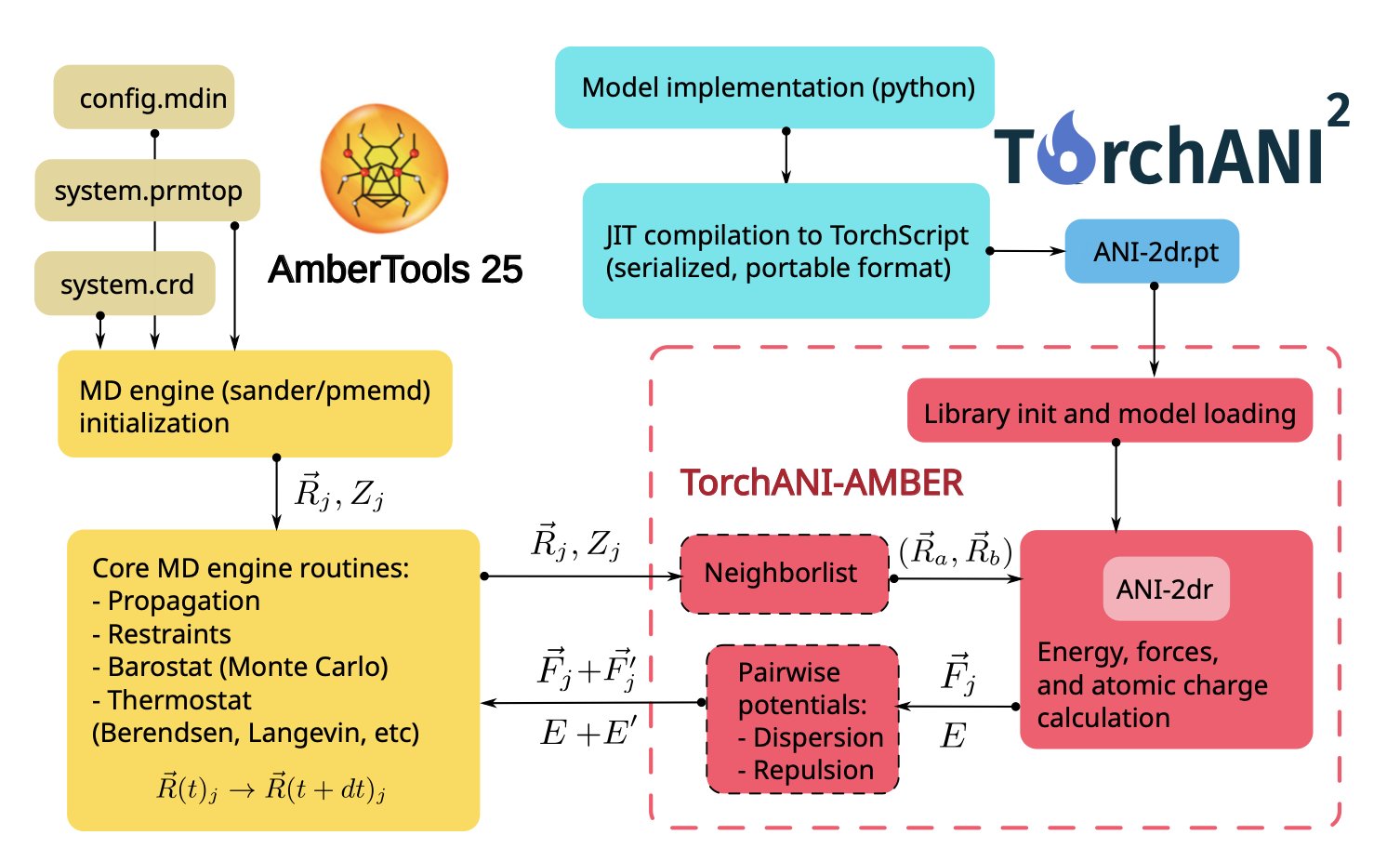
TorchANI-Amber,就是来打破这堵墙的。
核心思路:只搭桥,不拆庙
作者们没有重写 Amber 的底层代码,那将是一项浩大的工程。他们搭建一个外部接口。
这个接口的工作原理是这样的:
1. 在 Amber 的输入文件里,用户只需要做一个简单的设置,告诉 Amber:「这次模拟,不要用你自带的力场,请去问一个叫 TorchANI 的外部程序。」
2. 模拟开始后,Amber 在每一步,都会把当前体系所有原子的坐标,通过这个接口,发送给 TorchANI。
3. TorchANI 这边,是一个由 PyTorch 驱动的高效神经网络计算引擎。它接收到坐标后,会利用预训练好的 ANI 模型,快速计算出每个原子的能量和受力。
4. 然后,它再把这些计算好的能量和力,通过接口传回给 Amber。
5. Amber 拿到这些「外包」回来的结果后,就用它们来更新原子的速度和位置,继续下一步的模拟。
整个过程中,Amber 还是那个我们熟悉的 Amber,它负责处理积分、控温、控压、周期性边界条件等所有 MD 模拟的「杂务」。而最核心的、也是最耗时的能量和力计算,则被外包给了更专业的、精度更高的 AI 模型。
效果如何?是「花瓶」还是「利器」?
作者用一系列真实的、复杂的生物分子体系,证明了这套「混搭」系统不仅能跑,而且跑得很好。
TorchANI-Amber 的工作,为想用最先进的 AI 力场,去研究真实生物问题的研究者,提供了一极具示范意义的解决方案,在经典的、久经考验的软件框架上,通过巧妙的「嫁接」,可以催生出强大的新能力。
📜Paper: https://doi.org/10.26434/chemrxiv-2025-j0b7s