
目录
- 通过用化学上合理的片段替换分子的一部分,这个新方法让 AI 的预测解释,对药物化学家变得有用且可指导。
- PROTAC-Splitter 采用双模型混合策略,实现了 PROTAC 分子的自动化可靠拆分,为大规模数据分析与分子设计铺平了道路。
- AI 在化学领域的突破,是一场新旧工具并存的缓慢进化,核心是从手工构建特征转向 AI 自主学习。
1. AI 解释终于学会了做化学
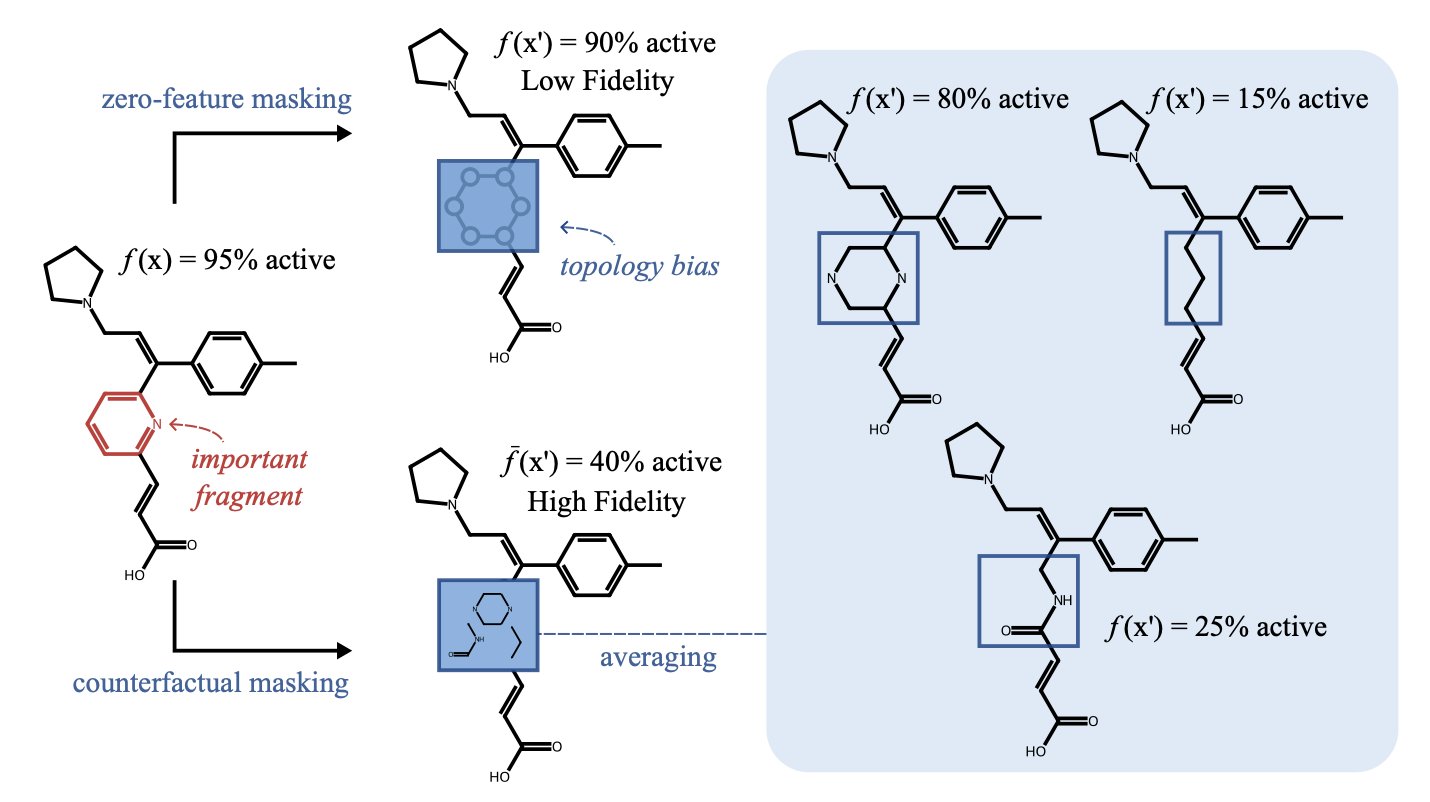
过去的 AI 解释方法,通过删除原子来判断其重要性,创造出化学上不存在的分子,其解释对化学家没有实际意义。新方法不再删除,而是替换。它用一个化学上合理的片段,换掉分子的一部分,再询问 AI 的判断,这个过程模拟了药物化学家的真实思考方式。
一线药物化学家对 AI 模型的感觉很复杂。你给它一个分子,它能迅速预测其溶解度、毒性或活性。但当你问为什么,AI 给出的解释却很笨拙。
它会用一种叫「特征归零」的方法,直接抹掉分子的一部分。比如,它会移除一个苯环,然后说:「去掉苯环后,溶解度变好了,所以这个苯环不好。」
这好比一个汽车工程师,为了说明引擎的重要性,把引擎从车里拆出来,然后指着那堆无法动弹的金属说:「看,没有引擎不行吧?」这种解释,对想改进引擎的人没有帮助。化学家无法处理一个被凭空删掉苯环的分子片段。
学会问一个更好的问题
一篇新论文让 AI 学会了提出化学家关心的问题。这个新方法叫 「反事实掩蔽」(Counterfactual Masking)。
它的思路从「如果我们删除这个基团会怎样?」,转变为「如果我们替换掉这个基团会怎样?」
它的工作方式如下:
当想知道分子上某个甲基为何重要时,新方法不会抹掉它。它会用一个生成式 AI 模型,构想出一些化学上合理的替代品,比如把甲基换成乙基、氯原子或羟基。
然后,它用这些新「合成」的、化学上真实存在的分子,询问预测模型:「这些新分子的性质有何变化?」
这正是药物化学家在实验室的工作方式。我们不会只盯着一个分子,我们会思考:「如果把这个甲基换成三氟甲基,亲脂性会改变,会不会和口袋里的氨基酸产生新的相互作用?」
这个新方法,将 AI 的解释语言与化学家的设计语言对齐了。
一种可指导的 AI 解释
AI 的解释由此变得可指导。
当 AI 建议把 A 基团换成 B 基团后,预测活性会提高,你就得到了一个可验证、并且化学上合理的下一步行动方案。它提供了一张能在实验室验证的合成路线图,而不是空泛的讨论。
研究者在多个数据集上验证了该方法。他们发现,这种方法生成的反事实分子,化学结构更合理,合成可及性也更高。
能完美预测一切的 AI 还很遥远。这个方法对计算资源的要求更高,生成式 AI 构想出的片段,也未必都容易合成。
📜Title: Enhancing Chemical Explainability Through Counter-flanking Masking
📜Paper: https://arxiv.org/abs/2508.18561
2. AI 精准拆解 PROTAC,解放药物化学家
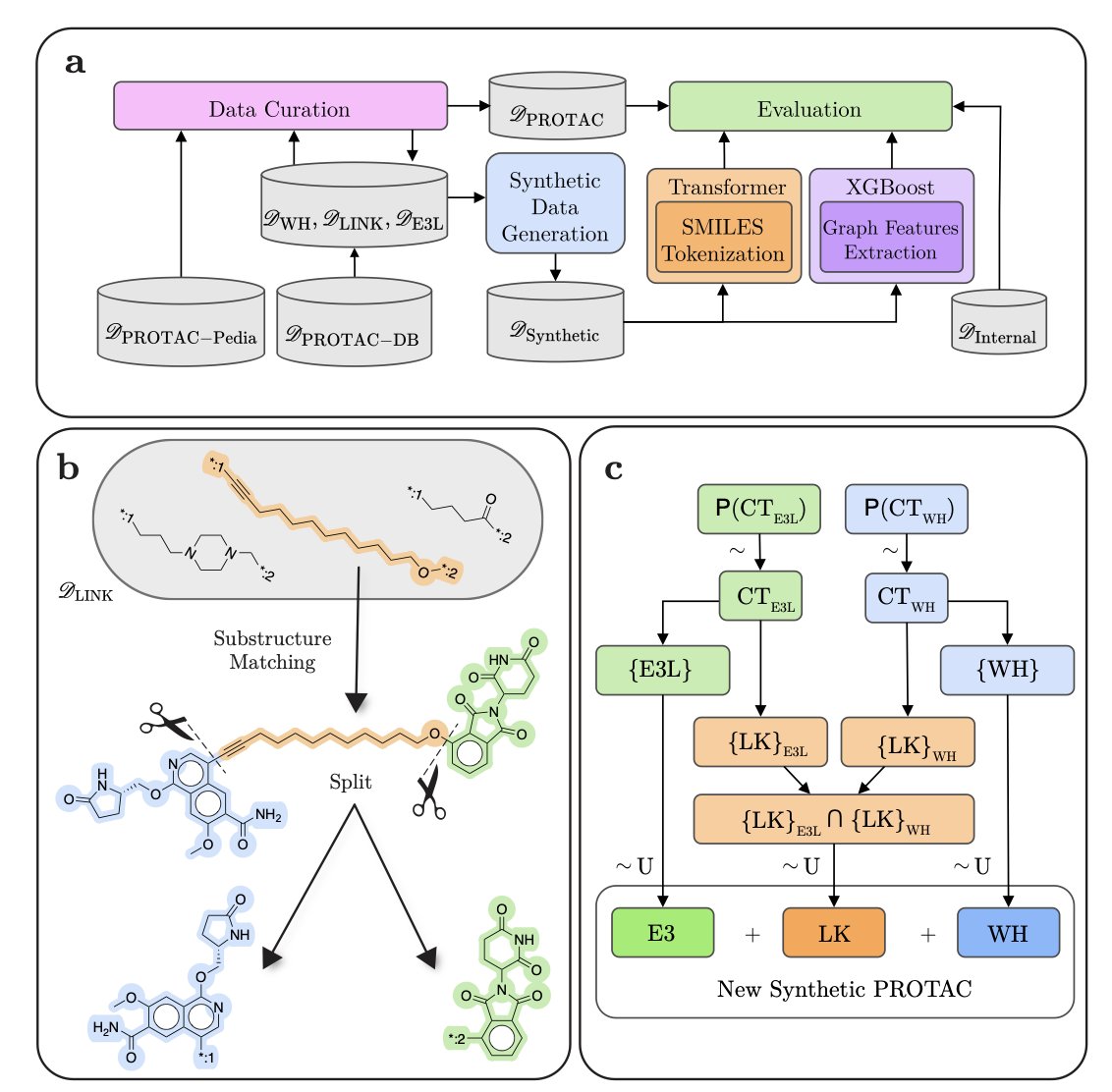
处理 PROTAC 数据库的研究者,都体会过手动将其拆分为弹头 (warhead)、接头 (linker) 和 E3 连接酶配体 (E3 ligase ligand) 的繁琐。这项工作不仅耗时,且容易出错,严重制约了新分子的分析与设计。一项新的自动化解决方案为此而生。
带标注的 PROTAC 数据稀缺,是机器学习方法遇到的首要障碍。为此,研究者主动出击,生成了 130 万个带详细注释的 PROTAC 分子。这批数据不仅用于训练模型,其本身也成为开放给整个社区的宝贵资源。
有了数据,他们设计了一套组合方案。首先是 Transformer 模型,它精度高,在公开数据集上表现出色。但作为生成式模型,它偶尔会产生化学上不合理的结构,导致拆分后的片段无法复原。
因此,他们还开发了一个基于图的 XGBoost 模型作为补充。该模型能 100% 保证化学合理性,确保拆分出的片段可以精确重构为原始分子。在药物研发中,可靠性至关重要。
这两个模型的结合方式是这套方案的关键。研究者设计了一套混合策略:系统优先使用 Transformer 模型进行拆分。只有当其结果未能通过化学有效性检查时,系统才会自动切换到 XGBoost 模型。这个纠错机制确保了最终输出结果的可靠性。
在药物研发的实际场景中,一个系统在追求高精度的同时,绝不能产出化学上无效的结果。PROTAC-Splitter 通过其混合策略实现了这一点。该工具已经开源,将帮助研究者们告别手动拆分 PROTAC 的低效工作。
📜Title: PROTAC-Splitter: A Machine Learning Framework for Automated Identification of PROTAC Substructures
📜Paper: https://doi.org/10.26434/chemrxiv-2025-bn1nv
💻Code: http://github.com/ribesstefano/PROTAC-Splitter
3. AI 化学突破:我们到底走到了哪一步?
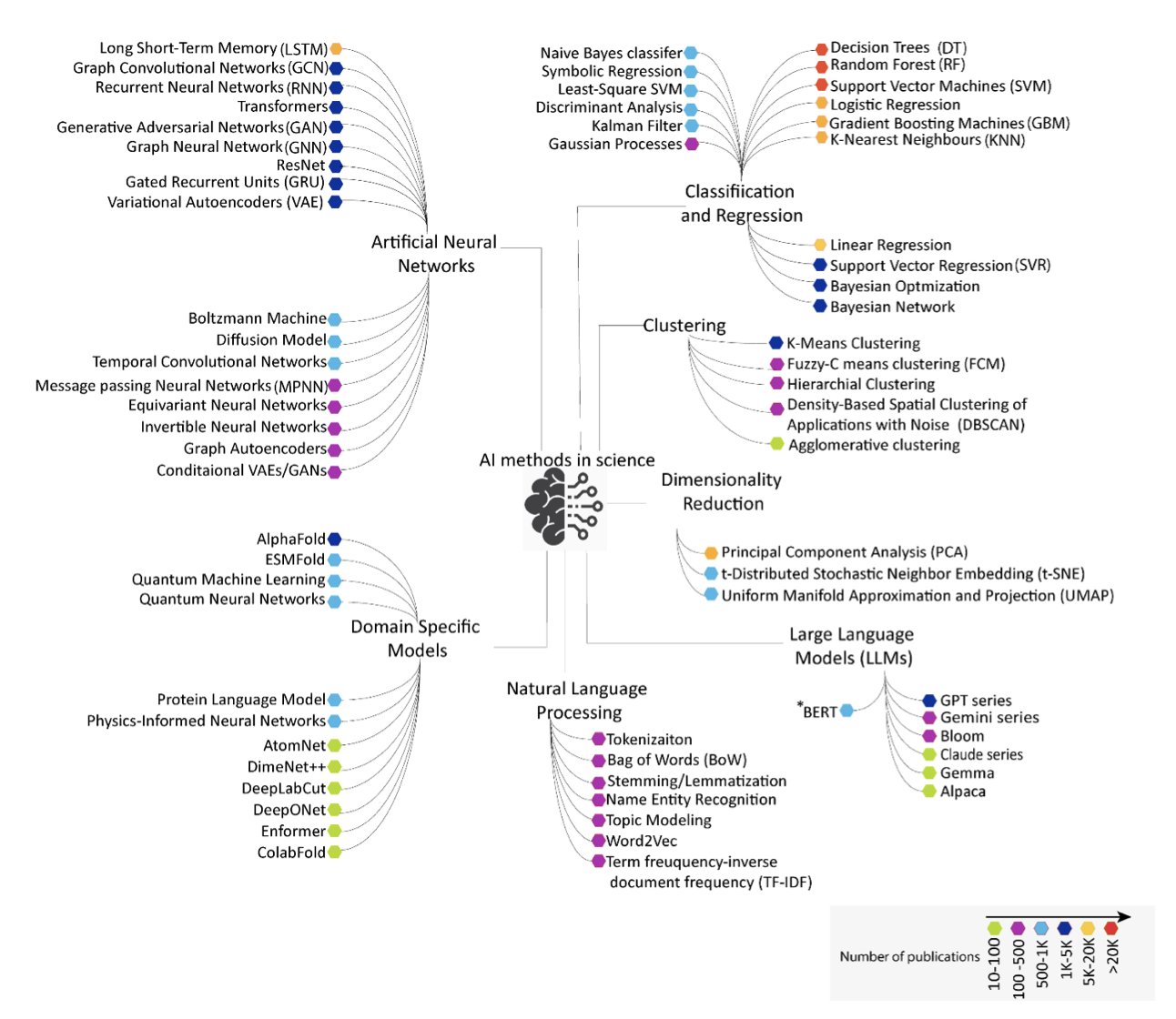
每隔几年,总有新技术宣称要让化学家失业。这次是人工智能。一篇综述分析了过去十年超过三十万份论文和专利,为这场技术变革描绘了一幅详尽的地图。
地图显示,真实情况比预想的更复杂,也更有趣。
老兵不死,只是在默默干活
随机森林、决策树等传统机器学习方法并未过时。在许多任务上,它们仍是最可靠的工具。
这些方法根植于化学家积累的化学直觉。我们将这些直觉量化为数千个「分子描述符」——例如分子的脂水分配系数、拓扑极性表面积等,再将这些精心准备的特征「喂」给模型。
这种「基于描述符」的方法让我们大致能理解模型的工作原理。
新贵登场,以及它带来的新问题
2020 年后,图神经网络 (GNNs)、生成模型等复杂的神经网络架构开始普及。
这些新工具引发了端到端学习的转变。
我们不再需要手工准备分子描述符,而是直接将分子的二维或三维结构交给 AI,让它自主学习需要关注哪些特征。这是一个飞跃。
大语言模型 (LLMs) 的加入,进一步拓展了应用边界。它们不仅预测分子性质,还在改变科研范式——从自动阅读海量文献、提出科学假说,到规划实验步骤。
最大的挑战:AI 的「黑箱」
AI 正从一个我们能理解其规则的工具,转变为一个能力强大但决策过程不透明的「黑箱顾问」。
它或许能提出卓越的设计方案,却无法解释原因。这种「不可解释性」在要求决策可追溯的严谨科学领域,是一个悬而未决的挑战。
这篇综述也提到了其他难题:高质量数据的稀缺、数据隐私,以及如何量化 AI 预测的「不确定性」。
所以,AI 化学的突破,目前尚未取代化学家。
它更像一场工具的进化。我们正从一个规则尚可理解的世界,进入一个由我们不完全理解的机器书写新规则的世界。
📜Title: The AI Revolution in Chemistry: Shaping the Future of Materials and Biomedical Sciences
📜Paper: https://doi.org/10.26434/chemrxiv-2025-8lrf0