
目录
- 一个自主 AI 智能体从零开始设计双靶点抗生素,展示了对抗超级细菌的全自动化流程。
- 我们终于有了一种能说化学母语的数学工具,它让 AI 不再将分子看作一堆原子,而是看懂了它们之间的连接关系。
- IDFlow 将物理能量作为 AI 的「导航」,引导它从盲目模仿转向有目的地寻找最稳定、最真实的 3D 结构。
1. AI Agent 的「双重打击」对付超级细菌
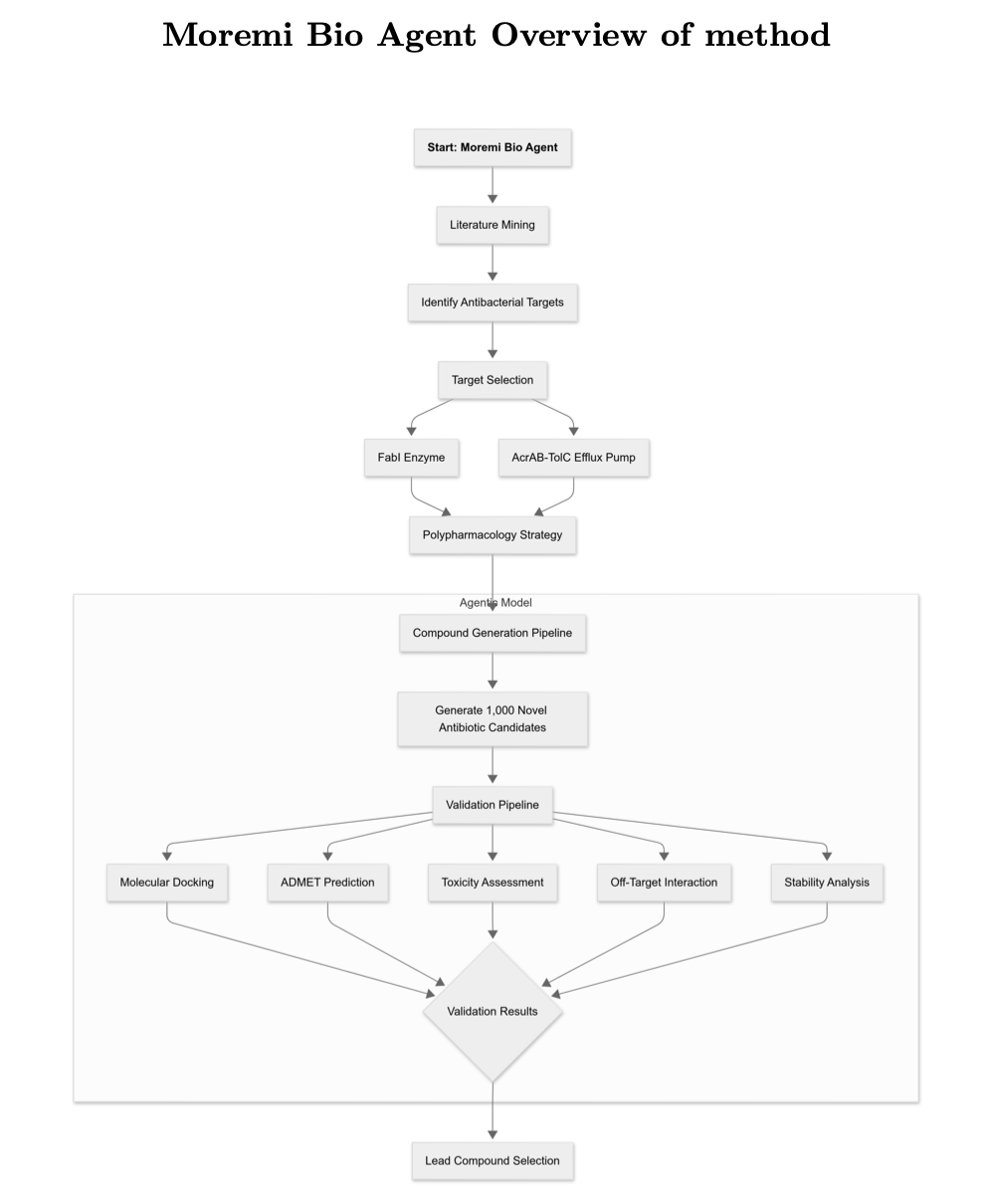
在抗生素研发领域,人类已屡屡受挫数十年。每当新药问世,肺炎克雷伯菌等革兰氏阴性菌总能迅速找到破解方法。这场对抗就像一场永无休止的「打地鼠」游戏,而我们手中的工具正日渐稀少。
一个公认的有效策略是:不要只攻击一个目标。假如能同时摧毁敌人的兵工厂和逃跑路线,胜算便会大增。
AI 的「钳形攻势」
研究者选择的两个靶点是 FabI 酶与 AcrAB-TolC 外排泵。FabI 酶是细菌合成细胞膜的「生产线」上的关键机器,AcrAB-TolC 外排泵则是细菌排出药物分子的「逃生通道」。
同时攻击这两个靶点,形成了一次「钳形攻势」。
战略确定后,执行任务交给了 AI。
自主的 AI 项目经理:Moremi Bio Agent
这项工作的主角是一个名为 Moremi Bio 的 AI 智能体。它并非简单的生成模型,而是一个全自动的项目经理。
它的工作流程如下:
第一步,生成。设计 1002 个理论上能同时抑制两个靶点的新分子。
第二步,筛选。对这 1002 个分子逐一进行分子对接、ADMET(吸收、分布、代谢、排泄,Absorption, Distribution, Metabolism, and Excretion)和毒性预测。
第三步,排序。根据类药性指标,为通过初筛的分子打分排序。
整个过程完全自主。
AI 的成果
1002 个初始分子中,774 个通过了初步的 ADMET 基准测试。最终,AI 项目经理提交了一份包含 60 个分子的候选短名单。
分析显示,其中 391 个分子对两个靶点都表现出「中等」强度的结合作用。
但「中等」这个词需要客观看待。
AI 尚未能一步到位设计出可直接进入临床的纳摩尔级药物。这 60 个分子更像是一批优质的先导化合物,是宝贵的研发起点。
这正是这项工作的价值。
传统药物发现中,从零开始寻找具备双靶点活性和良好类药性的起点,一个团队可能需要耗费一年甚至更久。
Moremi Bio 在计算机中高效完成了这个困难的、从 0 到 1 的探索。
这项工作展示的是一个能解决实际难题的工程方案。它提供了一个更优的研发起点,让研究人员能集中精力与资源,将这些有潜力的分子优化成真正的药物。
📜Title: Moremi Bio Agent: Leveraging Agentic Large Language Model for the Discovery of Broad-Spectrum Antibiotics for Enterobacteriaceae
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.21.671656v1
2. 图神经网络:化学的通用语言
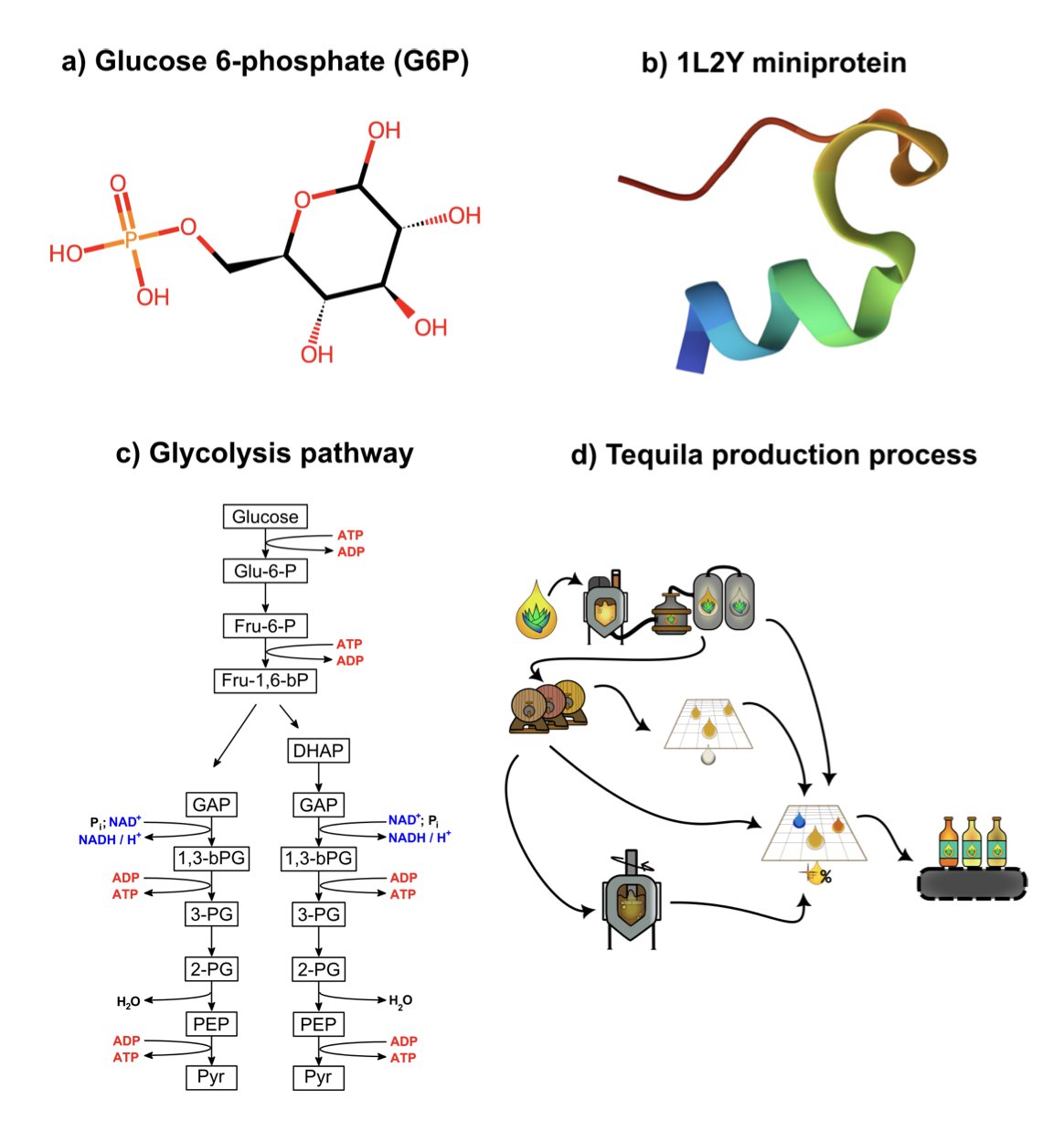
几十年来,我们试图教计算机化学,过程好比通过电话向一位盲人描述法拉利。你可以列出所有参数——四个轮子、两个门、一台 V12 发动机、红色车身,但对方永远无法理解这台机器的精髓:所有零件如何以优美而高效的方式连接在一起。
过去的机器学习方法正是如此。我们为每个分子计算上百个「分子描述符」,如分子量、脂水分配系数、环的数量、氢键给体与受体数。这本质上是一张详尽的「零件清单」。我们将清单喂给 AI,期望它能领悟化学原理。AI 有时能猜对,但它并未真正理解。
这篇综述宣告,一个新时代或许已经到来。我们找到了一种更好的语言与 AI 交流化学。
欢迎来到图的世界
这个新语言是图 (Graph)。在数学上,图的定义很简单:一堆节点和连接节点的边。
再看化学分子,它正是一堆原子(节点)和连接原子的化学键(边)。
这种对应堪称天作之合。
当我们将分子表示成图,我们不仅告诉 AI「这里有一个碳原子,那里有一个氧原子」,更重要的是,我们告诉它:「这个碳原子和那个氧原子连在一起。」
图神经网络便是为解读这种图语言而生的 AI。
它的工作方式符合化学直觉。信息在相连的原子间传递。一个碳原子会把它「我是 sp2 杂化碳,连接着一个氧和一个氮」这类信息传递给邻居。邻居收到信息后,再传递给自己的邻居。经过几轮信息传递,分子中的每个原子都对自身所处的化学环境有了丰富而深刻的认知。
这就是 GNN 在预测分子性质时表现出色的原因。它不再基于零散的「零件清单」猜测,而是在理解了整个分子的「设计蓝图」后做出判断。
不只是分子
这种方法的优美之处在于其普适性。
一个葡萄糖分子是一个图。整个糖酵解通路也是一个图,只是此时,图的节点变成了代谢物,边变成了催化这些转变的化学反应。我们可以用完全相同的数学框架,去研究完全不同尺度的问题。
这篇综述也展望了未来的方向:能理解三维空间信息的 3D 图、能描述化学反应过程的时间依赖图,以及将药物、靶点和疾病连接起来的庞大知识图谱。
图神经网络并非又一个时髦词汇,它代表一种转变。我们终于停止将化学硬翻译成计算机能处理的长串数字,而是开始教计算机学习化学家自己的母语——结构与连接。
📜Title: Graph Data Modeling: Molecules, Proteins, & Chemical Processes
📜Paper: https://arxiv.org/abs/2508.19356
3. AI 生成分子新招:用物理能量导航
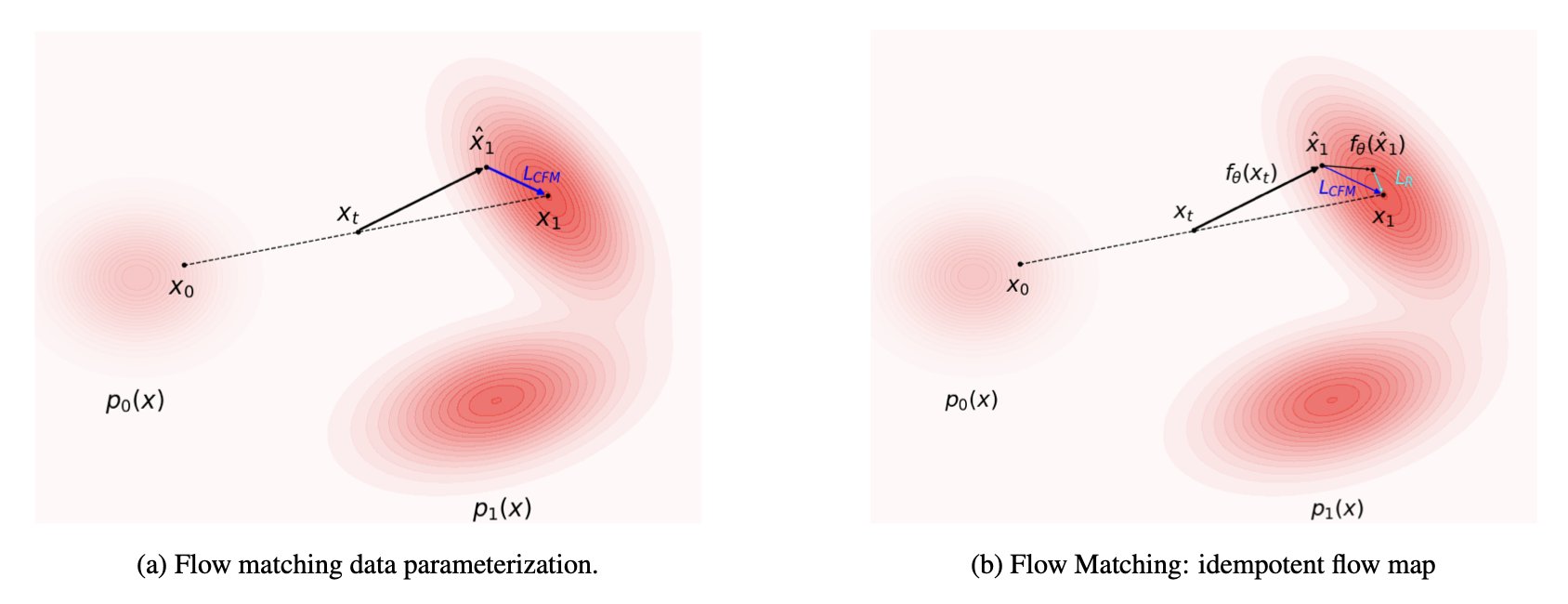
在 AI 生成分子的世界里,我们一直有个疑问:AI 到底是在「创作」还是在「抄袭」?
许多生成模型,例如扩散模型或流匹配模型,就像一个天赋异禀但有些莽撞的艺术家。你给它看一万张椅子照片,它就能画出第一万零一张看起来很像椅子的新椅子。但你问它,这把新椅子人坐上去会不会散架?它不知道。因为它只懂美学,不懂力学。
在分子设计这个严肃的工程领域,仅仅「长得像」远远不够。一个分子必须符合物理和化学的基本定律,能量上稳定,且能在真实世界里存在。否则,AI 生成的就成了「科学幻想画」,而非分子。
IDFlow 的工作原理,就像给这位艺术家 AI 请了一位严格的物理学导师。
让 AI 的目标函数更「物理」
IDFlow 的核心是一个简单的改动:它改变了 AI 的「学习目标」。
传统的流匹配模型(flow matching model),其学习目标是让生成结果在统计分布上逼近训练数据。
IDFlow 的目标则改为,让生成分子的「能量」尽可能低。
这里的「能量」被定义为模型的「重建误差」。在生成分子的过程中,AI 会不断进行「猜测」和「修正」。IDFlow 的训练过程,就是强迫 AI 的每一次修正,都朝着降低分子整体能量、优化结构合理性的方向进行。
这好比教建筑师画图纸。过去只要求图纸与范例相似,现在要求每一稿都通过结构力学软件的稳定性测试。这样,最终的图纸自然兼具美观与可行性。
结果怎么样?
当 AI 开始尊重物理学,它交出的作业质量就上了一个台阶。
研究者在两个一线研发人员最关心的任务上,检验了 IDFlow 的性能:
第一个是分子对接。结果显示,IDFlow 在预测药物分子与靶点蛋白结合方式时,准确度(RMSD 指标)超过了现有的基准模型。
第二个是蛋白质骨架生成。这是一个更难的任务,相当于让 AI 从零开始设计一个全新的蛋白质。结果,IDFlow 生成的蛋白质骨架具有更高的「可设计性」,意味着更有可能为这个骨架找到能稳定折叠成它的氨基酸序列。它生成的骨架摆脱了空有其表的「花架子」。
实现这一切并未带来巨大的额外计算成本。
这项工作让 AI 从一个「模式模仿者」向真正的「物理世界模拟器」迈出坚实一步。它没有试图用一个黑箱去解决所有问题,而是将我们熟知的基本物理学原理,融入 AI 的学习过程。
📜Title: Energy-Based Flow Matching for Generating 3D Molecular Structure
📜Paper: https://arxiv.org/abs/2508.18949