
目录
- MLFGNN 通过融合图注意力网络、图 Transformer 和分子指纹,试图同时捕捉分子的局部细节和全局结构,在基准测试中表现亮眼,但能否在真实的药物研发项目中炼出真金,还有待检验。
- MolSnap 用一种新的、几乎瞬时完成的生成方法,直接挑战了当前又慢又贵的扩散模型,而且生成的分子质量还更高。
- PocketXMol 用一种极其底层的原子级视角,打造了一个真正意义上的「一个模型通吃」的分子设计平台,并且用湿实验结果证明了它不是在玩票。
- PepThink-R1 通过让大语言模型学习「思考链」(Chain-of-Thought),在优化环肽时不仅能给出结果,还能解释「为什么这么改」,让 AI 的决策过程不再是黑箱。
- PromptBio 是一个 AI 驱动的「生物信息学部门 in a box」,它能自动处理从标准化多组学分析到自然语言定制流程的所有事情,直击该领域可重复性的核心痛点。
1. MLFGNN:图神经网络的「远近视」矫正术
一个分子的性质,到底是取决于某个特定的官能团,还是它整体的拓扑结构?
答案是「都重要」。
这就给图神经网络 (GNN) 出了个大难题。多数模型要么擅长聚焦局部,像戴着一副高倍放大镜,能把一个酯基或一个卤素看得清清楚楚;要么擅长鸟瞰全局,能把握整个分子的形状和柔性。想让一个模型同时拥有这两种「视力」,太难了。
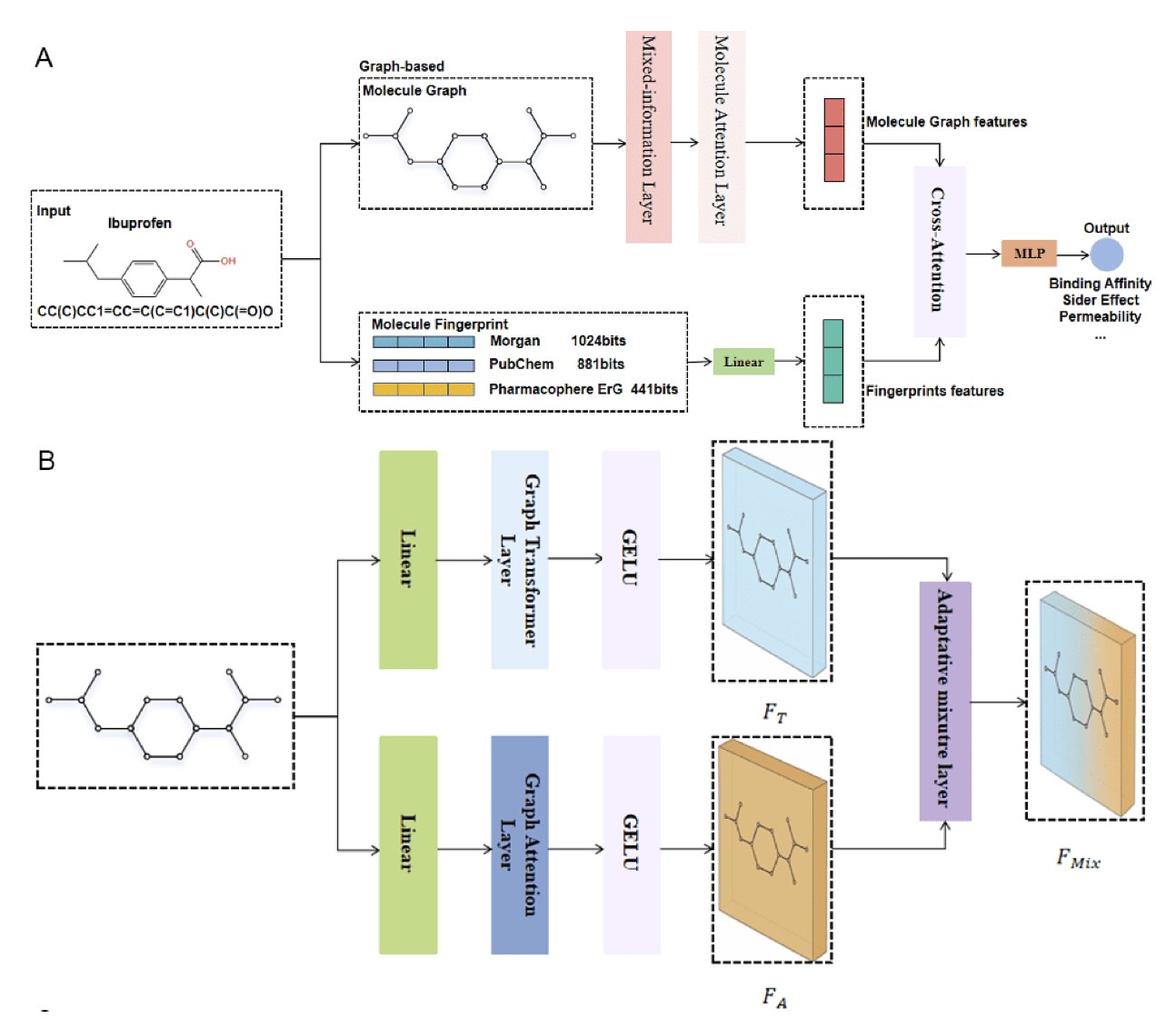
MLFGNN,用图注意力网络 (GAT) 来处理局部信息。你可以把 GAT 想象成一队嗅探犬,对每个原子周围的化学环境极其敏感。这是模型的「近视」能力,负责识别那些决定局部反应性或结合模式的关键基团。
接着,为了获得「远视」能力,他们引入了一个图 Transformer。Transformer 这东西在自然语言处理里大杀四方,就是因为它擅长捕捉长距离依赖关系。在这里,它被用来审视整个分子图,理解那些跨越多个化学键的相互作用,比如分子的整体偶极矩或是复杂的立体效应。
最妙的地方在于那个所谓的「自适应融合机制」。这就像一个智能的自动对焦系统。如果模型要预测的性质(比如水溶性)更依赖分子整体的极性表面积,系统就会给全局信息(Transformer 的输出)更高的权重。反之,如果要预测某个位点的代谢稳定性,那局部化学环境(GAT 的输出)就更关键。这个机制让模型自己学会了「看情况办事」,而不是一刀切。
还没完。研究者们把传统的分子指纹也整合了进来。分子指纹是化学信息学里的老将了,简单、快速,凝聚了几十年的化学知识。MLFGNN 用一个交叉注意力模块,让 GNN 学到的新潮特征和这些经典指纹特征进行「对话」,互相取长补短。这感觉就像让一个天赋异禀的年轻化学家去请教一位经验丰富的老法师,避免了 GNN 在复杂的图空间里「想太多」而钻牛角尖。
论文里的基准测试结果,一如既往地漂亮。在好几个公开数据集上,MLFGNN 都把现有的一流方法甩在了身后。但这毕竟是在「驾校」里考试。真正的挑战是把这辆车开上真实药物研发项目这条「野路」。真实世界的分子库充满了各种偏见和从未见过的骨架。这个精巧的「混合动力」系统能否经受住考验,还是会因为过于复杂而过拟合?
代码已经开源,现在整个社区都可以把 MLFGNN 拉出来遛遛,用自己手里那些乱七八糟、充满挑战的专有数据去检验它。只有这样,我们才能知道,这个新模型究竟是一个能在炼丹炉里点石成金的法宝,还是又一个在基准测试里闪闪发光、一到实战就熄火的「银样镴枪头」。
📜Title: Multi-Level Fusion Graph Neural Network for Molecule Property Prediction
📜Paper: https://arxiv.org/abs/2507.03430v1
💻Code: https://github.com/lhb0189/MLFGNN
2. AI 制药一键生成:MolSnap 告别慢速扩散模型
如果关注 AI 药物发现,你一定对「扩散模型」(Diffusion Models)这个词不陌生。
它们是当下的宠儿,能生成非常高质量的分子。但它们有一个致命的弱点:慢。慢得让人发指。它们就像一个细心的雕刻家,从一块随机的「噪音」大理石开始,一刀一刀,极其耐心地凿出最终的分子形状。这个过程,可能需要几百甚至上千步。
这当然很优雅,但在快节奏的药物研发世界里——结果可能不错,但黄花菜都凉了。
MolSnap 冲着扩散模型喊话:「你们太慢了!」
它的核心武器有两个,一个比一个狠。
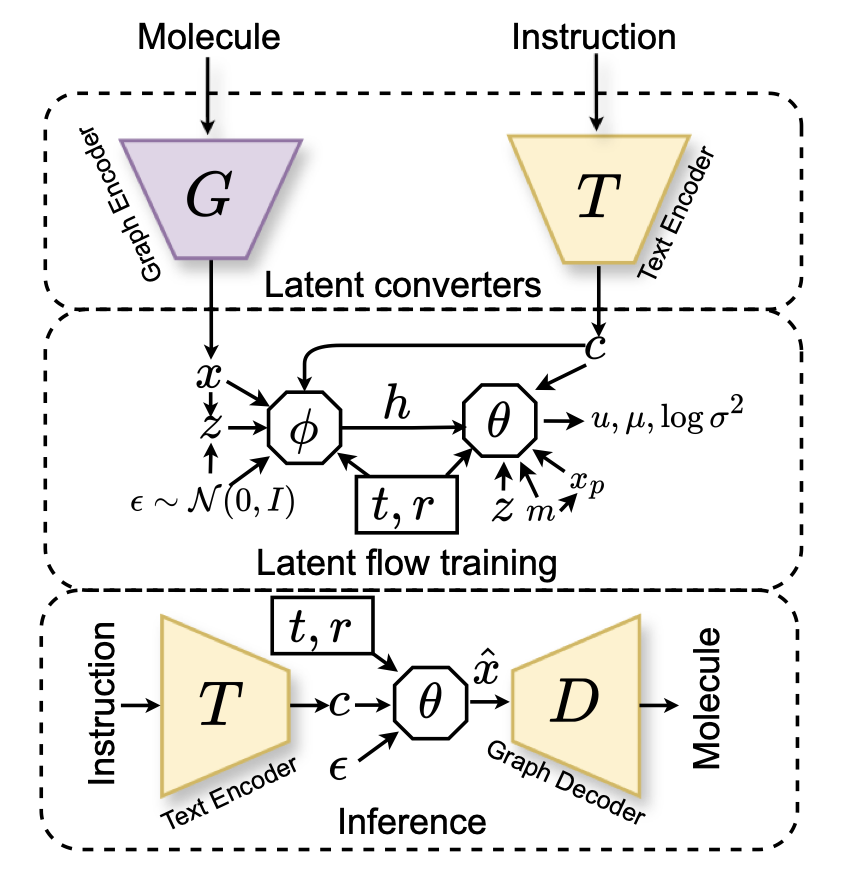
第一个叫「变分均值流」(VMF)。你不需要理解它背后的复杂数学。你只需要知道,它让分子生成,从一个「千步走」的过程,变成了一个「一步到位」的过程。它不像扩散模型那样亦步亦趋地从噪音走向分子,而是直接学习了从起点到终点的「路径图」,然后一步就跳到了终点。论文说它只需要 1-5 次函数评估就能搞定,这在计算效率上,简直是把自行车换成了喷气机。
第二个,也是保证质量的关键,叫做「因果感知 Transformer」(CAT)。这解决了另一个大问题。很多模型在根据文本(比如「我想要一个能抑制激酶、水溶性好的分子」)生成分子时,就像是在玩文字拼图,把各种特征硬凑在一起,但不管它们之间的化学逻辑。而 CAT 在生成时,会强制执行「因果依赖」。这就像你搭乐高,你必须先放好底座,才能在上面放砖块。这种对化学构建逻辑的尊重,直接带来了一个惊人的结果:100% 的化学有效性。它不会再给你生成那些五配位的碳或者其他让有机化学家血压飙升的怪物。
所以,MolSnap 不仅快,而且质量高。它生成的分子在新颖性和多样性上,都比现有最好的方法要强。
我们可以用一种交互式的、几乎没有延迟的方式去探索化学空间。我们可以像和聊天机器人对话一样,快速地测试各种「如果……会怎样」的想法。「如果我想要一个有三个氢键供体,cLogP 小于 2 的分子会是什么样?」MolSnap 几乎可以立刻给你答案。
📜Title: MolSnap: Snap-Fast Molecular Generation with Latent Variational Mean Flow
📜Paper: https://arxiv.org/abs/2508.05411v1
3. AI 制药:这次真做出了活性分子
我们的电脑硬盘里,塞满了各种各样的 AI 药物设计工具,简直像一个杂乱的动物园。有一个专门生成小分子的,有一个专门设计多肽的,还有一个专门搞对接的……它们各自为政,互不相通。你每次想干点新活,就得找个新工具,然后学习它那套独特的「方言」。
PocketXMol 有种秦始皇搞「书同文、车同轨」的感觉。
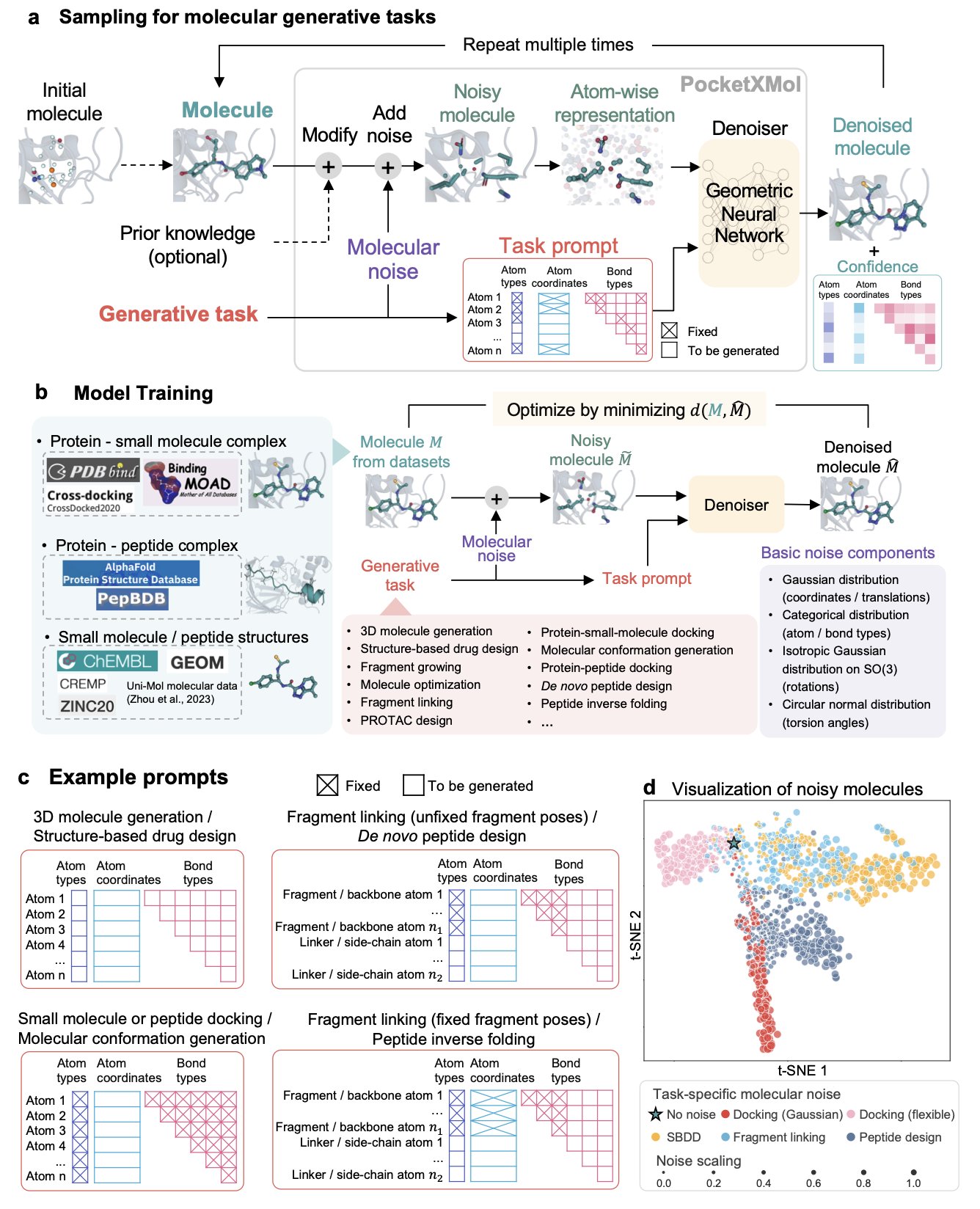
PocketXMol 说别再分什么小分子、多肽了,这些都是人为的概念。在最根本的物理层面上,它们是什么?它们不就是一堆原子,以特定的三维方式排列在蛋白质的口袋里吗?
于是,PocketXMol 就用这种最底层的、最不带偏见的「原子级」视角,去训练一个巨大的基础模型。它不学习「化学」,它学习的是「原子该如何被放置」。这就像你教一个 AI 当厨师,不是给他一百万份菜谱去背,而是直接教他热力学、分子美食学和美拉德反应的原理。
它的训练方式,是现在很流行的「从噪音中恢复结构」,这很像扩散模型。但它的应用方式,是「任务指令」。你不需要为每个新任务去痛苦地微调模型。你只需要像跟 ChatGPT 聊天一样,告诉它:「嘿,给我设计一个能结合到这个口袋里的小分子」,或者「给我设计一条能塞进这个槽里的多肽」。它就能听懂,然后开始在原子层面「创作」。
好了,理论说完了,我只关心一件事:它管用吗?
他们不只是在基准测试上跑了个高分(虽然它确实在 13 个任务里拿了 11 个第一),他们真的下场了。
* 他们让 PocketXMol 去设计 Caspase-9 的抑制剂。结果,AI 设计出的新分子,活性和市售的抑制剂相当。
* 他们让 PocketXMol 去设计 PD-L1 的结合肽。结果,成功率远超随机筛选。
能同时在小分子和多肽这两个规则迥异的赛场上取得成功,这极大地证明了它那个「原子级」统一表示法的威力。AI 不需要知道什么是氨基酸,什么是苯环,它只需要知道,在特定的物理化学环境下,哪些原子的组合是最稳定的。
📜Title: PocketXMol: An Atom-level Generative Foundation Model for Molecular Interaction with Pockets
📜Paper: https://www.biorxiv.org/content/10.1101/2024.10.17.618827v2
💻Code: https://github.com/pengxingang/PocketXMol
4. PepThink-R1:会「思考」的环肽优化 AI
现在用大语言模型(LLM)来设计多肽,已经不是什么新鲜事了。我们给模型一个初始的环肽序列,让它去优化,它确实能吐出来一个新序列,各项预测指标也都不错。但你问它:「你为什么要把第 5 位的丙氨酸换成一个非天然氨基酸?」它答不上来。
这种「知其然,而不知其所以然」的黑箱模式,让我们很难完全信任 AI 给出的设计。我们不知道它做出这个修改,是真正学到了底层的化学规律,还是仅仅拟合了训练数据里的某些巧合。
PepThink-R1 框架让 LLM 在做环肽优化时,不仅能干活,还能「写工作日志」。
怎么教 AI「写工作日志」?
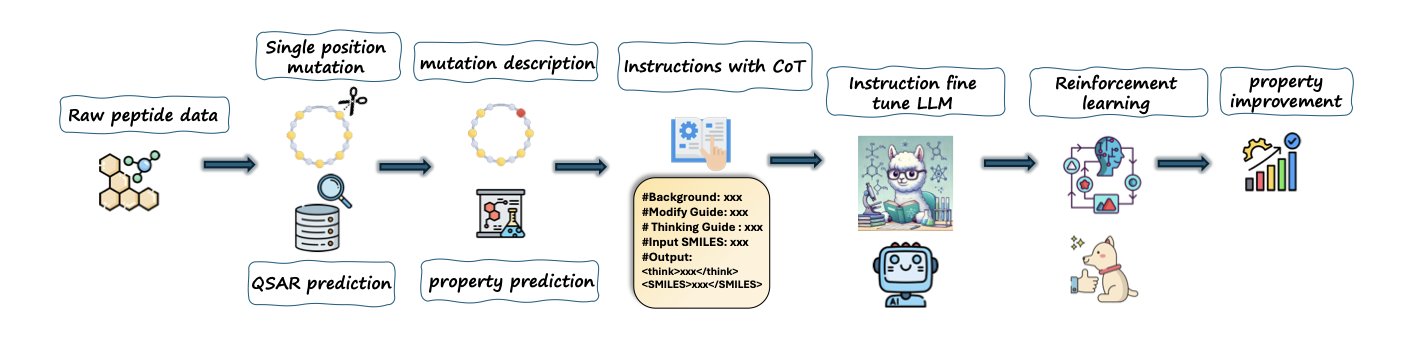
核心武器是现在很火的「思考链」(Chain-of-Thought, CoT)技术。
作者构建了一个全新的训练数据集,每一对「优化前 - 优化后」的环肽序列,都配上了一段「设计思路」作为标签。
这个「设计思路」会明确地写出:
他们用这些带有「工作日志」的数据,对 LLM 进行监督微调(SFT)。这就相当于,手把手地教模型,如何把一个具体的氨基酸修改,和我们期望达到的药学性质改进,这两件事给关联起来。
从「学会思考」到「自主探索」
当模型通过 SFT,初步掌握了这种「思考 - 修改」的模式后,PepThink-R1 会进入第二阶段:强化学习(RL)。
在这个阶段,模型会开始自主地去探索各种各样的氨基酸修改方案。它每做出一次修改,都会得到一个「奖励分数」。这个分数会综合评估新生成环肽的化学合理性、以及各项性质(脂溶性、稳定性等)的改善程度。
通过不断地试错,模型会逐渐学会,什么样的「操作」能获得高分,什么样的修改是「瞎折腾」。
这个「SFT + RL」的两步走策略,给了模型一个很好的起点,让它不至于在强化学习的巨大探索空间里迷路;而 RL 则给了模型超越训练数据的能力,让它能发现一些全新的、更优的解决方案。
结果如何?
在和通用 LLM 以及领域内专门的基线模型 PepINVENT 的对比中,PepThink-R1 在优化成功率和结果的可解释性上,都表现出了明显的优势。它的高质量成功率(HQSR)达到了 0.890,这意味着它给出的建议,绝大多数都是靠谱的、有价值的。
最关键的是,它生成的那些「思考链」,本身就是宝贵的信息。它能启发我们从新的角度去思考构效关系,让 AI 真正成为我们研发工作中的一个可以「对话」的、智能的合作伙伴。
📜Paper: https://arxiv.org/abs/2508.14765v1
5. PromptBio:你的 AI 生物信息学专家团队
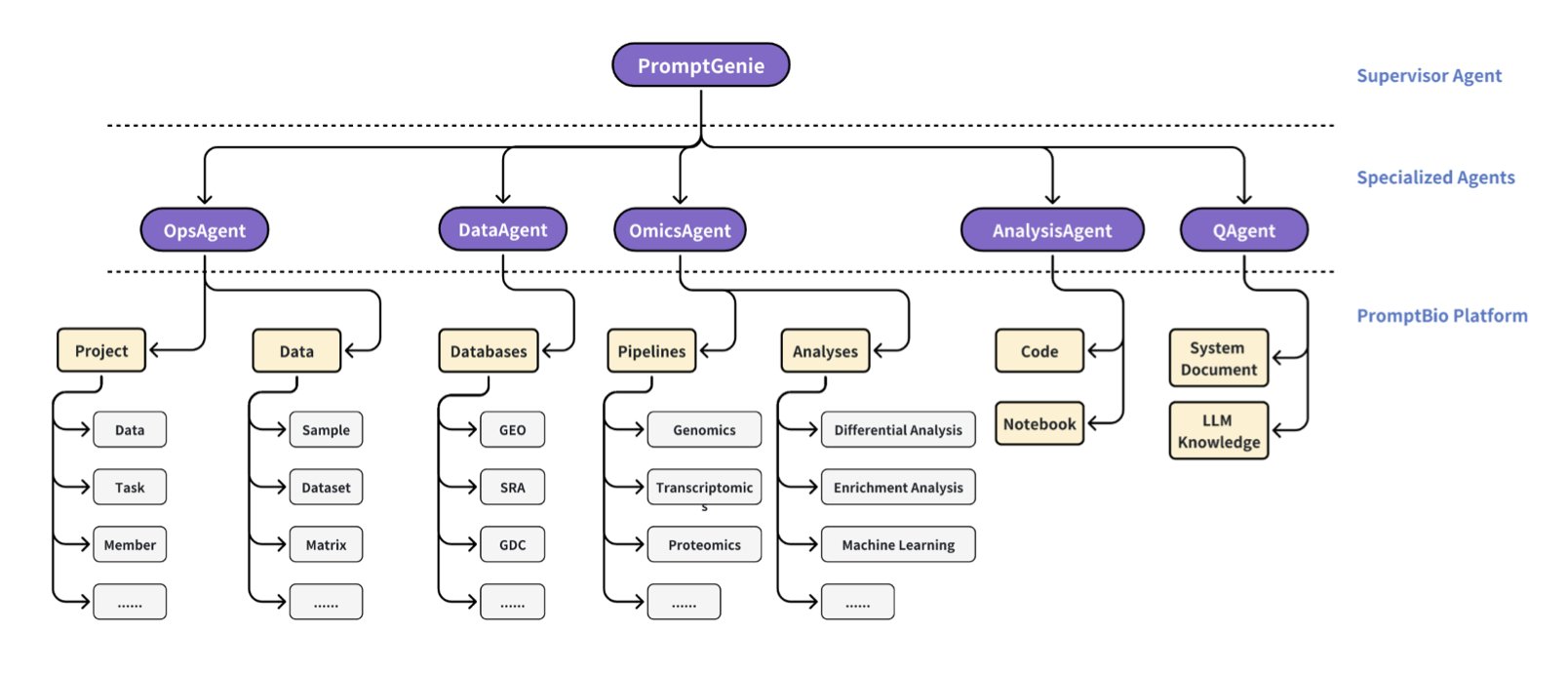
生物信息学分析,是现代生物医学研究的引擎,也是无数研究生的噩梦:一堆来源不明的脚本,一个永远也配不平的环境,还有那句经典的「在我的电脑上是能跑的」。六个月后想重复一下自己当初的分析,发现比登天还难。这个领域充满了手工作坊式的操作,可重复性简直是一场灾难。
PromptBio 这个平台,就是想把我们从这场灾难中解救出来。它不是又一个孤零零的分析工具,它的架构是一个「多智能体系统」,说白了,就是给你配了一个 AI 团队,有总管,有干活的专员,各司其职。
这个团队里有几个明星员工:
第一个叫DiscoverFlow。你可以把它想象成一个极其严谨、甚至有点强迫症的「流水线工人」。你给它一个标准的多组学分析任务,它会严格按照一个预设的、不可更改的流程图(DAG)来执行。好处是什么?绝对的可重复性。今天跑和明天跑,你跑和我跑,结果一模一样。对于大规模、标准化的数据处理,这就是我们梦寐以求的。
第二个叫ToolsGenie,这是团队里的「创意天才」。你不用再学那些复杂的命令行了。你直接用大白话跟它说:「嘿,帮我找出我的肿瘤样本里,那些过表达的、同时又是激酶的基因。」ToolsGenie 就会自己去挑选合适的工具、生成脚本、把它们组装起来,然后给你答案。它把你从「怎么做」的繁琐细节里解放出来,让你专注于「做什么」和「为什么」的科学问题。
第三个叫MarkerGenie,这是团队的「图书管理员」和「文献分析师」。当你从前两个模块拿到一份基因列表时,你是不是还得自己吭哧吭哧地去 PubMed 上一个一个搜,看它们是不是已知的生物标志物?MarkerGenie 帮你干了这个活。它会自动去文献库里检索,然后给你一份总结报告,告诉你这些基因在疾病中扮演过什么角色。
PromptBio 从一开始就把安全和合规放在了台面上。加密、权限控制、审计日志,瞄准 HIPAA 和 SOC2 标准。这清楚地表明,它的目标是进入真正的临床应用,处理敏感的病人数据。这和那些只在学术圈里自娱自乐的项目,完全是两个维度的思考。
📜Paper: https://www.biorxiv.org/content/10.1101/2025.07.05.663295v1