
目录
- CBYG 框架通过贝叶斯流网络和梯度引导,为结构化药物设计提供了一种比扩散模型更稳定、更可控的 3D 分子生成方法。
- 在分子语言模型领域,盲目追求大模型和大数据集并非正途,MolEncoder 证明了更优的预训练策略才是关键。
- DeepSEA 是一个可解释的深度学习模型,它能绕过序列比对的限制,更准确地识别新型抗生素耐药蛋白,并揭示其作用机制。
1. CBYG:超越扩散模型,3D 分子生成迎来新范式
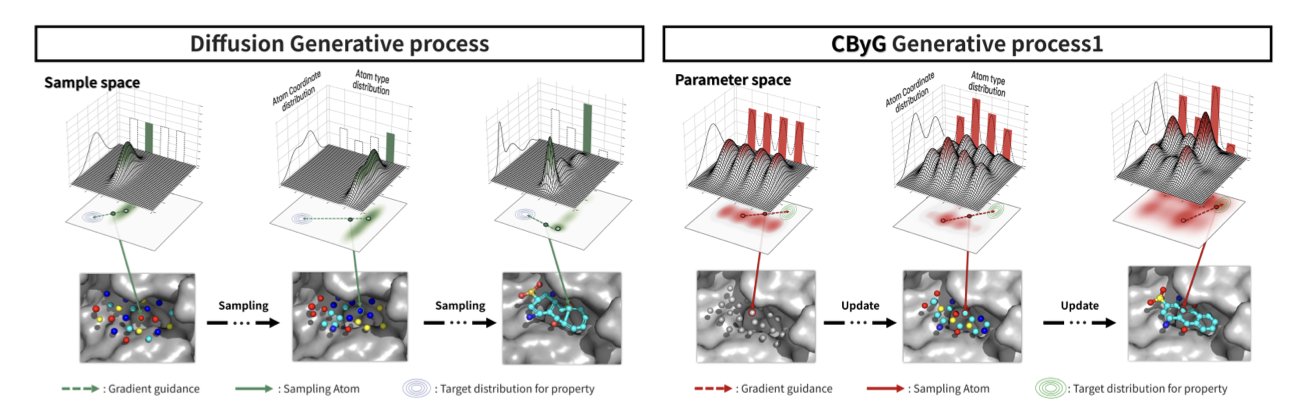
做 AIDD 这些年,我们见证了 AI 生成模型的爆发。特别是基于结构的药物设计,扩散模型 (Diffusion Models) 几乎成了标配。但只要你亲手跑过这些模型,就会知道它们有多「任性」。你想要一个高亲和力的分子,它可能会给你一个化学上根本不存在的结构。这背后的核心难题,在于模型很难同时处理好两件事:原子的三维空间位置(连续坐标)和原子的类型(离散的化学元素)。它们就像两个配合不佳的舞伴,步调总是不一致。
这篇来自 Choi 等人的新工作,提出的 CBYG 框架,就是来解决这个问题的。他们采用了贝叶斯流网络 (Bayesian Flow Networks, BFNs)。
我们可以这样理解两者的区别。扩散模型的工作方式,好比从一团随机的「原子云」(噪声)开始,一步步把它「去噪」,最终雕琢出一个分子。这个过程很强大,但每一步的修正都可能引入不确定性,尤其是在施加外部引导(比如「提高亲和力」)时,整个系统容易崩溃。
而 BFNs 的工作原理更像是在搭积木。它有一个更清晰、更有序的路径,从一个简单的初始状态,通过一系列可学习的「流动」变换,逐步构建出最终的复杂分子。这个过程的稳定性天然就更好。
CBYG 的精髓在于,它在 BFNs 这个稳定的底盘上,加装了一个精准的「导航系统」——梯度引导。在分子生成的每一步,这个系统都会根据我们设定的目标(比如结合亲和力、合成可行性、选择性)计算出一个「梯度」,然后像施加一个微小的力一样,把生成过程往正确的方向推一把。
打个比方,这就好比我们不是在对一块大理石进行大刀阔斧的雕刻,而是在用 3D 打印机逐层构建。我们可以在打印的每一层都进行微调,确保最终的产品完全符合设计图纸。这种方式让整个生成过程变得可控。当你想让分子更「好」一点时,模型不会给你一个完全离谱的东西,而是在现有基础上稳步优化。
更让我欣赏的是研究者们设计的评估体系。很多 AI 论文热衷于在一些学术指标上刷分,但这些指标和我们制药工业的日常需求相去甚 - 远。这次,他们把重点放在了结合亲和力、合成可及性分数 (SA score) 和选择性这些我们真正关心的「硬指标」上。实验结果显示,CBYG 在这些贴近实战的基准测试中,性能超过了现有的主流模型。
对于我们来说,它不再是一个充满惊喜(和惊吓)的「黑箱」,而是一个可以信赖的、能听懂我们化学直 - 觉的工程系统。CBYG 当然不是终点,但它指明了一个有吸引力的方向:让 AI 分子生成变得更像一门严谨的工程科学,而不是一门玄学。
📜Title: Controllable 3D Molecular Generation for Structure-Based Drug Design Through Bayesian Flow Networks and Gradient Integration
📜Paper: https://arxiv.org/abs/2508.21468v1
2. MolEncoder:分子预训练,告别「越大越好」
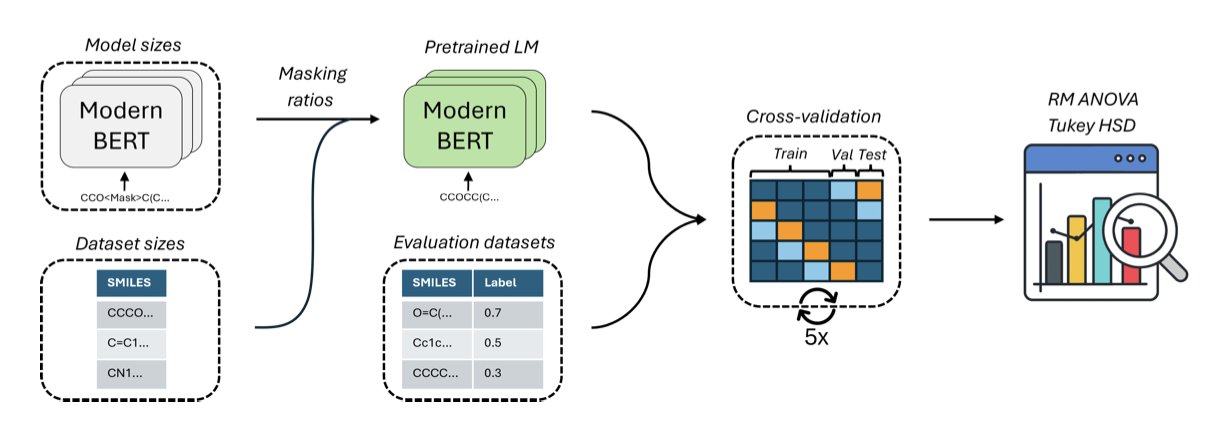
近年来,AI 领域被一个观点主导:模型越大、数据越多,效果就越好。从 GPT 到图像生成模型,这条「力大砖飞」的路线似乎百试百灵。药物发现领域也尝试借鉴,开发了基于分子 SMILES 序列的「分子语言模型」。但分子与语言不同,直接照搬自然语言处理(Natural Language Processing, NLP)的最佳实践,效果并不理想。
这篇新研究表明,这种照搬是行不通的。
这些模型的工作原理,是把分子的 SMILES 字符串看作一个句子,如CCO(乙醇)。预训练的核心是「遮蔽语言建模」(Masked Language Modeling),即玩填字游戏。随机盖住句子里的某些部分,让模型去猜。例如将CCO变为C[MASK]O,模型需根据上下文猜出被盖住的是C。
在 NLP 领域,公认的黄金法则是遮蔽 15% 的词。但研究者发现,这套规则用在分子上效果不佳。分子的语法比人类语言严格得多。在英语里,将”a quick brown fox”换成”a fast brown fox”,意思相近。但在化学里,将乙醇(CCO)的一个碳换成氮(CNO),就变成了完全不同的物质。
因此,研究者尝试提高遮蔽率。他们发现,当遮蔽率提高到 30% 时,模型在下游任务(如预测分子性质)上的表现大幅提升。这相当于给模型出了更难的考题,迫使它不再是死记 SMILES 的局部模式,而是去理解构成一个稳定、有效分子的深层化学规则。
另一个发现关乎模型和数据规模。我们通常认为模型参数越多、见过的分子越多,它就越「博学」。但实验结果显示,一个仅有 1500 万参数的中等大小模型,在使用一半 ChEMBL 数据集进行预训练时,表现最佳。继续增大模型尺寸或喂给它全部数据,性能反而开始下降。
这说明,对于分子性质预测这类任务,存在一个「甜点区」。超过这个区域,更大的模型可能会学到噪音,或者说,更多的参数和数据带来了「收益递减」。这对于计算资源有限的实验室是好消息,我们无需追逐动辄数十亿参数的巨型模型,也能做出顶尖成果。
基于这些发现,作者构建了 MolEncoder。它是一个在「甜点区」里精心调校过的 BERT 模型。在多个行业标准测试集上,这个小巧的 MolEncoder 全面或部分超越了更大、更复杂的模型,如 ChemBERTa-2 和 MoLFormer。它用更少的计算资源,完成了更出色的工作。
最后,这项研究还指出:预训练损失(pretraining loss)并不能很好地预测模型在实际任务中的表现。很多团队会盯着这个数字,认为它越低模型就越好。但事实是,必须将模型在真实的下游任务上进行测试,才能知道其真实性能。
📜Title: MolEncoder: Towards Optimal Masked Language Modeling for Molecules
📜Paper: https://doi.org/10.26434/chemrxiv-2025-h4w9d
3. DeepSEA:用 AI 揪出隐形的抗生素耐药蛋白
抗生素耐药性 (Antimicrobial resistance, AMR) 是一个日益严峻的问题。我们现在监控耐药性的主流方法,如 RGI 和 AMRFinderPlus,严重依赖序列比对。这就像手里拿着一本嫌疑人名单,然后去比对现场发现的指纹。如果罪犯是个新手,不在名单上,你就抓不到他。这些工具的假阴性率因此居高不下,一旦出现序列新颖的耐药蛋白,它们就会失灵。
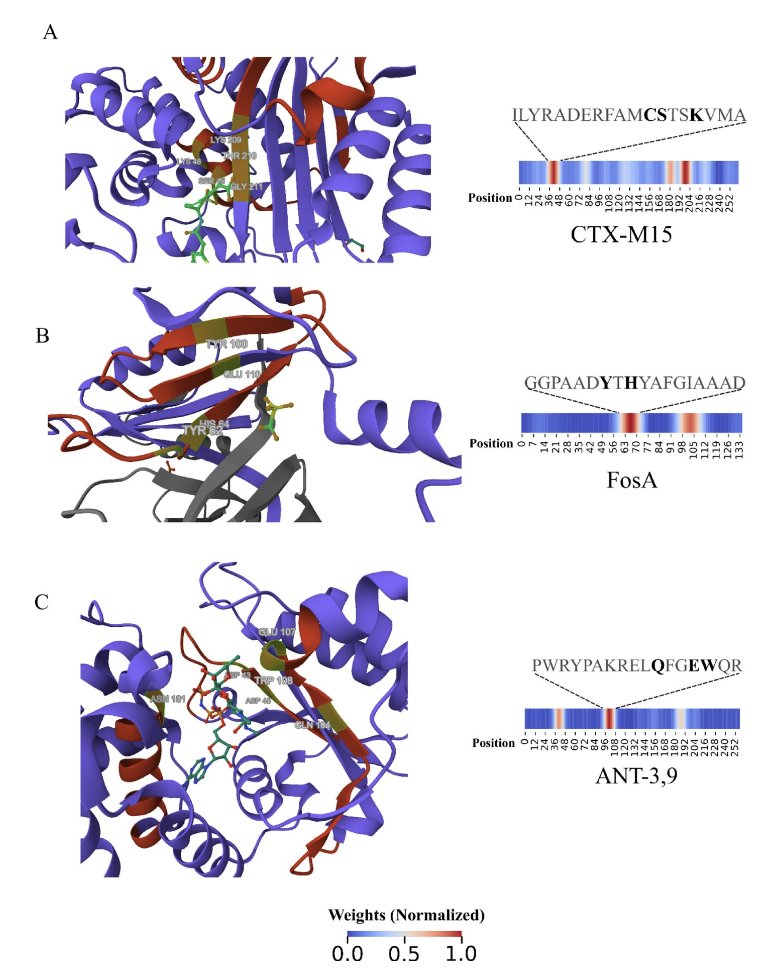
这篇论文里的 DeepSEA 模型,试图用一种新思路解决这个问题。它是一个卷积神经网络 (Convolutional Neural Network, CNN),本质上是一种模式识别工具。你可以把它想象成一个训练有素的专家,看的不是完整的序列,而是序列中的关键「片段」或「基序」。它不关心整个蛋白序列和数据库里的已知序列有多像,只关心它是否包含那些赋予其耐药功能的关键结构域。
这是它的工作原理。研究者用一个名为 NCRD 的非冗余综合数据库来训练模型。他们把大量的耐药蛋白和非耐药蛋白序列「喂」给 CNN。网络通过学习,自动提取出与耐药性相关的抽象特征。比如,某个特定的氨基酸组合可能形成了一个可以水解抗生素的活性位点。CNN 就能学会识别这个模式,即使它出现在一个全新的蛋白骨架上。
这个模型让我欣赏的一点是它的可解释性。我们最怕的就是「黑箱模型」。一个模型告诉我这个蛋白有耐药性,但说不出为什么,那它的价值就大打折扣。DeepSEA 的研究者通过分析 CNN 内部神经元的激活模式,反向追溯是输入序列的哪个部分让模型做出了「耐药」的判断。这样一来,模型不仅给出了预测,还用高亮的方式「指」出了蛋白上可能负责降解抗生素的关键功能域。这就从一个单纯的分类工具,变成了一个能启发新实验、帮助我们理解耐药机制的工具。
模型的性能数据也很扎实。在测试中,它对九类耐药蛋白的召回率 (Recall) 都超过了 0.95。这意味着它几乎能把所有真正的耐药蛋白都找出来,漏网之鱼很少。在和 RGI、AMRFinderPlus 甚至是 ESM2 这种大模型的对比中,DeepSEA 的假阴性更少。
研究者还测试了它的泛化能力。他们用一些和训练集序列相似度很低的蛋白去测试模型,结果模型的精确率、召回率和 F1 分数依然很高。这说明 DeepSEA 不是靠「死记硬背」来识别序列,而是真正学到了决定蛋白耐药功能的底层规律。
最后,作者们把这个模型打包成了一个开源的生物信息学工具,提供了命令行界面。这意味着任何一个实验室都可以下载下来,用于分析自己的宏基因组数据,去发现那些可能被传统方法忽略的新型耐药基因。
📜Title: DeepSEA: An Alignment-Free Explainable Approach to Annotate Antimicrobial Resistance Proteins
📜Paper: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06256-4
💻Code: https://github.com/computational-chemical-biology/DeepSEA-project