
Table of Contents
- ApexGen uses flow matching to simultaneously optimize peptide sequence and 3D structure in a single, deterministic process, solving the traditional problem of disconnected sequence and structure design to improve binding affinity and physical realism.
- The PRIMO framework combines in-context learning with test-time training to predict protein fitness, including complex insertion and deletion mutations, using only a small amount of experimental data.
- Traditional static models struggle to capture the transient interactions in gene regulation. This study proposes a new computational framework that integrates multi-omics data with temporal dynamics to solve this problem.
- Rep3Net fuses molecular descriptors, ChemBERTa, and graph features to accurately predict the activity of PARP-1 inhibitors, outperforming single-modality models.
- Using a fine-tuned ESM2, a two-layer prediction framework accurately identifies transcription factors and reveals their binding preferences for DNA methylation and key sequence motifs.
1. ApexGen: Simultaneously Generating Peptide Sequence and Structure with Flow Matching
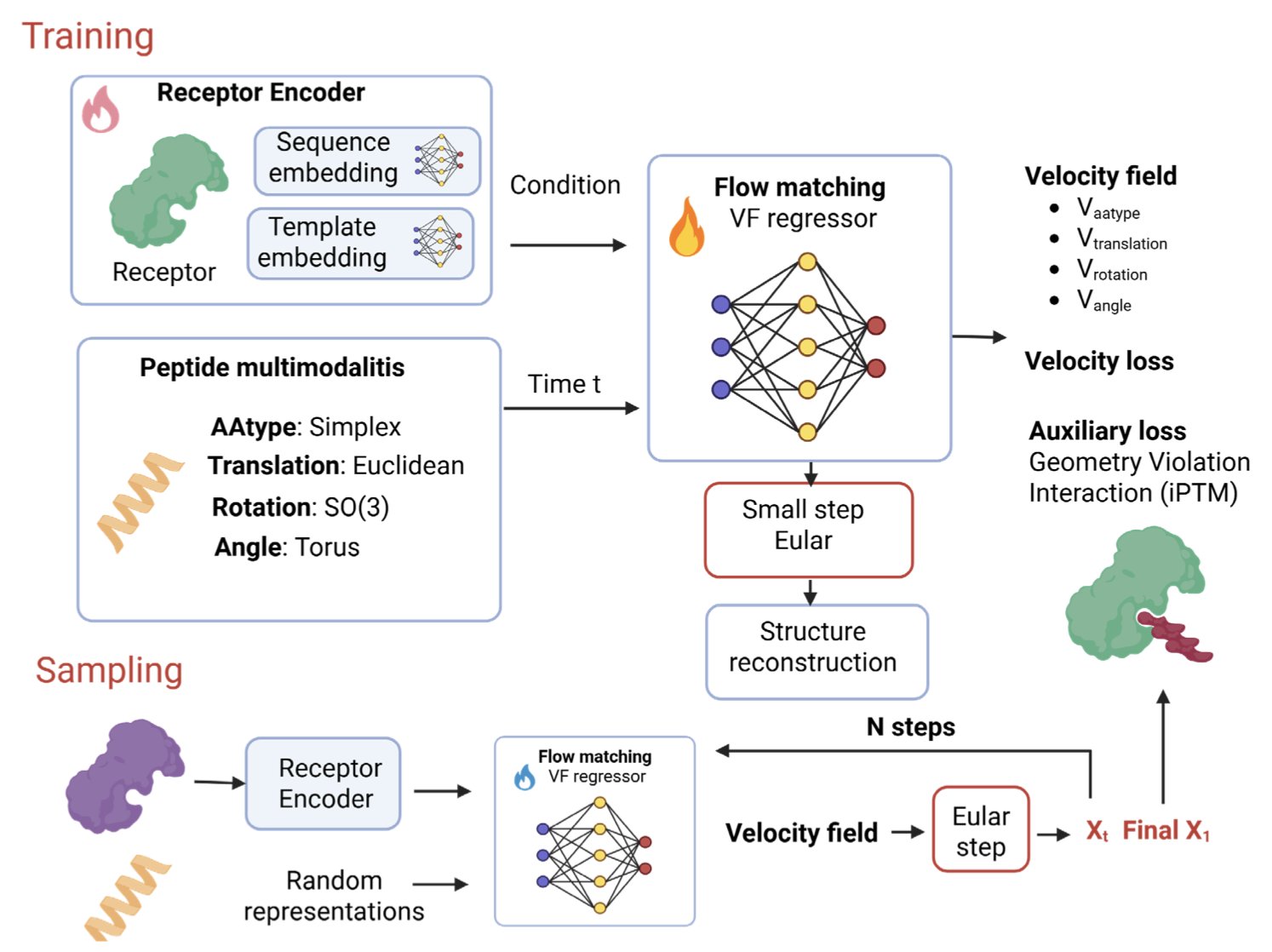
Peptide drug design has long been held back by computational complexity. Traditional methods usually treat “sequence optimization” and “structure generation” as separate steps. This approach often leads to a trade-off: a seemingly good sequence can’t fold into the right shape, or a perfect structure is chemically impossible to synthesize.
ApexGen offers a new AI framework to break this deadlock. It uses flow matching in a mathematical space called a “product manifold” to handle a peptide’s backbone torsion, rigid body position, amino acid type, and side-chain angles all at once. The peptide’s sequence identity and geometric structure evolve together in the same process.
The advantage of this method is geometric consistency. Unlike many models that generate structures needing a “fix” to obey physical laws, ApexGen produces structures that are physically plausible from the start, with no extra corrections needed.
In comparisons with existing models like PepFlow, ApexGen shows strong performance. Its generated structures are highly similar to naturally folded peptides, with an average TM-score of 0.99, which is close to the theoretical limit in computational structural biology.
Whole-receptor conditioning shows the depth of this model’s design. The AI no longer just focuses on the binding pocket but perceives the context of the entire receptor protein. This global view ensures the peptide fits precisely into the binding site and avoids steric clashes with other parts of the receptor.
From a chemical perspective, the generated peptides are highly realistic. They follow natural Ramachandran plot distributions, have natural backbone angles, and show good chemical intuition—they know to use polar and aromatic residues at key interfaces to enhance binding. Evaluations with Boltz-2 and PyRosetta also confirm that these designs are more energetically favorable.
ApexGen provides a powerful tool for tackling difficult protein-protein interaction (PPI) targets in drug development. It can quickly generate diverse and structurally sound candidate molecules, significantly shortening the early screening cycle. As the library of non-natural amino acids expands and experimental data is fed back into the loop, this tool will become even more useful.
📜Title: ApexGen: Simultaneous design of peptide binder sequence and structure for target proteins 🌐Paper: https://arxiv.org/abs/2511.14663
2. PRIMO: Solving Few-Shot Prediction in Protein Engineering
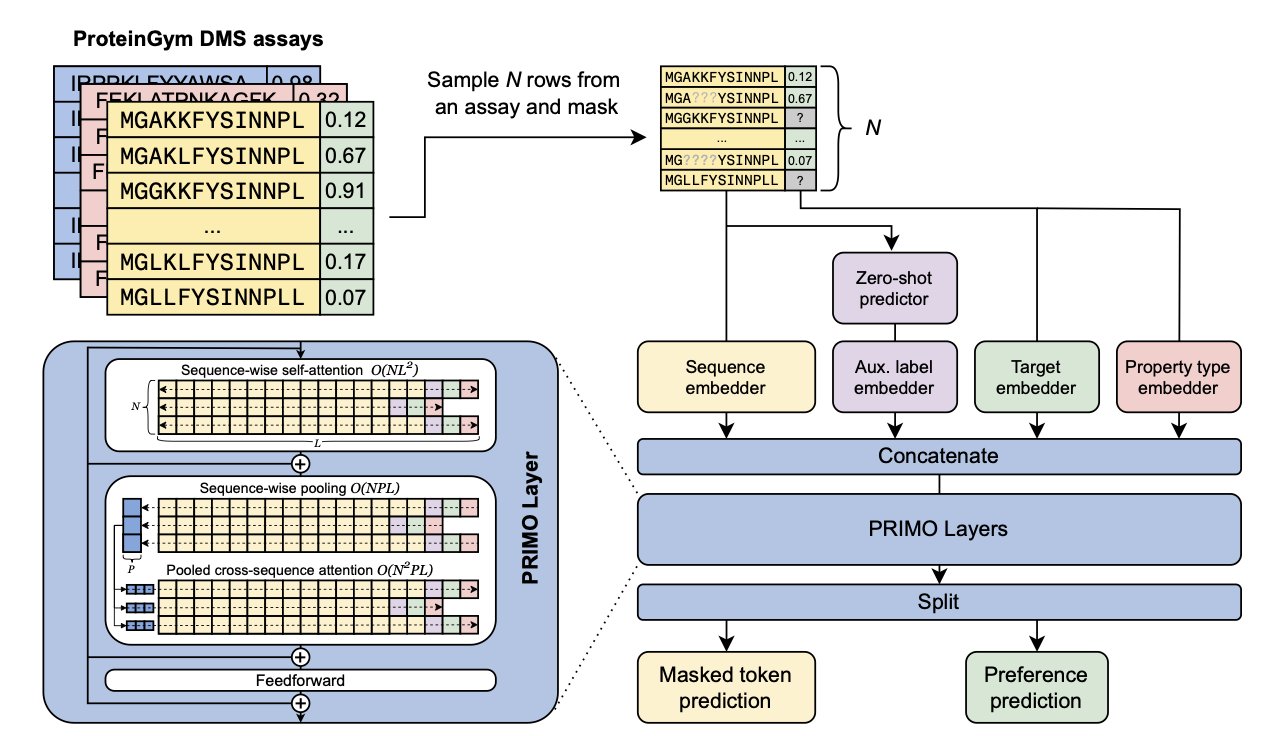
Protein and antibody engineering often faces a dilemma: you need a lot of data to predict the effect of a mutation, but you usually only have a handful of wet lab results. Deep learning models typically require massive datasets to train. Zero-shot models that rely on evolutionary information can run, but they struggle to capture the nuances of specific experimental conditions. The PRIMO framework was designed to solve this “few-shot” problem.
Core Logic: In-Context Learning and Test-Time Training
PRIMO uses an in-context learning approach similar to GPT: you show the model a few examples like “mutant A has high activity, mutant B has low activity,” and it learns to predict the activity of mutant C.
On top of this, PRIMO introduces test-time training. During the inference stage, the model quickly fine-tunes itself using the scarce experimental data available. It’s like cramming for a test by focusing on specific types of questions, adapting to the current prediction task.
How It Works
The researchers designed a unified encoding method to feed three types of information into the model: 1. Protein sequence information. 2. Auxiliary zero-shot prediction scores (baseline values calculated from existing evolutionary models). 3. Sparse experimental labels.
This information is converted into tokens and processed in a pre-trained masked-language modeling framework. To distinguish between good and bad mutations, the researchers use a preference-based loss function. The model focuses on the relative ranking of mutants rather than their exact values, which is more practical for screening highly active molecules.
Tackling “Insertion and Deletion” Mutations
Substitutions are relatively easy to handle. Insertions and deletions (indels), which change the length of the sequence, often cause alignment-based models to fail. PRIMO can effectively handle these complex changes to protein structure, showing strong robustness.
No “Gaming the Leaderboard”: More Realistic Benchmarking
Randomly splitting datasets can lead to overly high similarity between the training and test sets, inflating performance scores. The researchers introduced a “natural evolution benchmark” to ensure that training and test sequences are sufficiently different. The results show that even in this more challenging setup, PRIMO outperforms existing zero-shot and fully supervised benchmark models.
For drug development professionals who are working with a new target and only have a few dozen data points, PRIMO is a tool worth trying.
📜Title: Few-shot Protein Fitness Prediction via In-context Learning and Test-time Training 🌐Paper: https://arxiv.org/abs/2512.02315v1
3. Combining Multi-omics and Temporal Dynamics to Reconstruct Gene Regulatory Network Analysis
The field of computational biology has long worked to simulate Gene Regulatory Networks (GRNs). Old models were like static photographs—they could capture the relationships between molecules at a single moment but missed the process of their interaction. A new study has built a computational framework that strings these “photographs” together into a continuous “movie.”
Breaking Down Data Silos
The core of this framework is the integration of multi-omics data. Understanding complex cellular states requires this kind of holistic perspective. Biological processes are intricate, with genetic and epigenetic factors intertwined. The new framework reveals how different layers of regulators work together to influence gene expression.
Introducing the Time Dimension
Traditional analysis often ignores fleeting signals, but this research focuses on the temporal dynamics of gene regulation. During development or disease progression, key regulatory events are often transient. The new framework shows how these brief interactions can trigger a “butterfly effect,” leading to long-term changes in cell function. This ability to capture dynamic changes is crucial for understanding biochemical mechanisms.
From Simulation to Practice
An algorithm’s real-world use depends on its ability to handle data. The researchers optimized the framework’s scalability, allowing it to efficiently process terabyte-scale high-throughput sequencing data, which is essential for industrial applications. Validated through both simulated experiments and real biological scenarios (like developmental biology case studies), this tool is not just theoretical—it can uncover regulatory mechanisms that were previously overlooked.
📜Title: A New Approach in Computational Biology: Exploring the Dynamics of Gene Regulatory Networks 🌐Paper: https://arxiv.org/abs/2511.14676
4. Rep3Net: Fusing Multimodal Information to Accurately Predict PARP-1 Inhibitor Activity
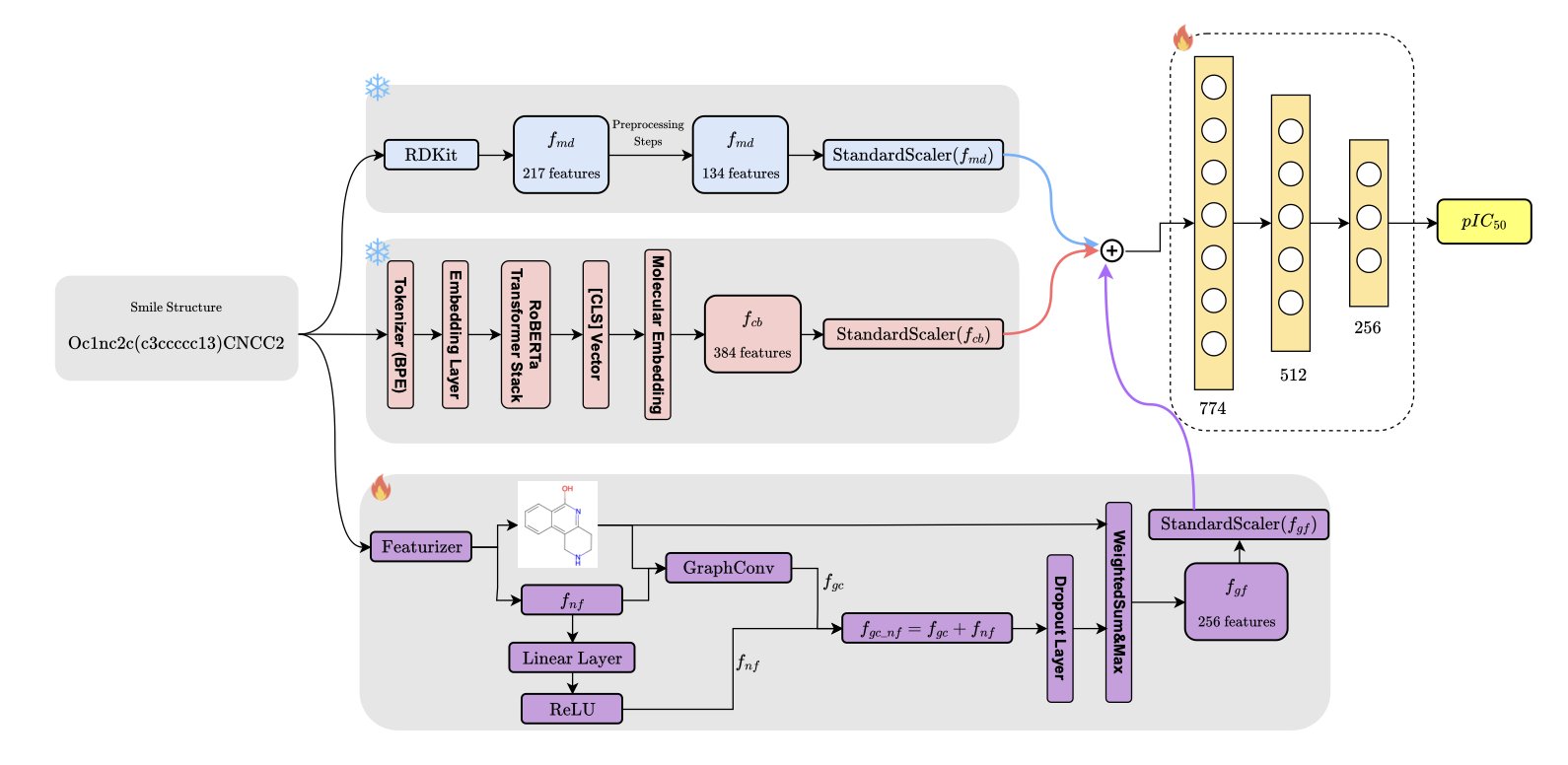
When predicting drug activity, researchers often choose between different methods: classic molecular fingerprints, Graph Neural Networks (GNNs), or Transformer-based Large Language Models (LLMs). This study on Rep3Net shows that fusing multiple methods is a better solution.
The Need for a Multimodal Perspective
Any single way of representing a molecule has its limits. Rep3Net’s architecture is designed to fuse three sources of information: 1. Molecular descriptors: These capture the physicochemical properties of a molecule, like molecular weight and LogP. 2. ChemBERTa embeddings: A pre-trained model based on Transformers that treats SMILES strings as a chemical language to extract contextual information. 3. Graph features: These directly process the molecule’s topological structure, focusing on the connections and spatial relationships between atoms.
This multimodal strategy is designed to make up for the shortfalls of any single approach. Data from an ablation study confirmed that removing any one module causes the model’s prediction performance to drop. All three together achieve the best results.
Performance in Action: PARP-1 Inhibitors
The researchers chose Poly(ADP-ribose) polymerase 1 (PARP-1) as their test target. PARP-1 is a well-established and important target in cancer therapy, with a large amount of high-quality activity data available, making it a convincing benchmark.
The results showed that Rep3Net performed better than traditional QSAR models on metrics like mean squared error, coefficient of determination, and Pearson correlation coefficient. This means its predictions and ranking ability are more reliable when screening for new compound scaffolds targeting PARP-1.
Balancing Computing Power and Engineering
Rep3Net has been optimized in its engineering implementation. It uses a parallel architecture to process the different molecular representations. This ensures the model is powerful enough to capture complex structure-activity relationships while maintaining reasonable computational efficiency. This is critical for early-stage drug discovery, which requires rapid screening of large virtual compound libraries.
What’s Next?
The model has mainly been validated on PARP-1. The future challenge is to generalize this architecture to other targets where the chemical space is broader and the data is more sparse. Also, multimodal models often face issues with interpretability. Using explainable AI techniques to clarify the basis for the model’s decisions will be crucial for earning the trust of chemists.
📜Title: Rep3Net: An Approach Exploiting Multimodal Representation for Molecular Bioactivity Prediction 🌐Paper: https://arxiv.org/abs/2512.00521v1
5. Fine-Tuning ESM2 to Accurately Identify Transcription Factors and Their Methylation Binding
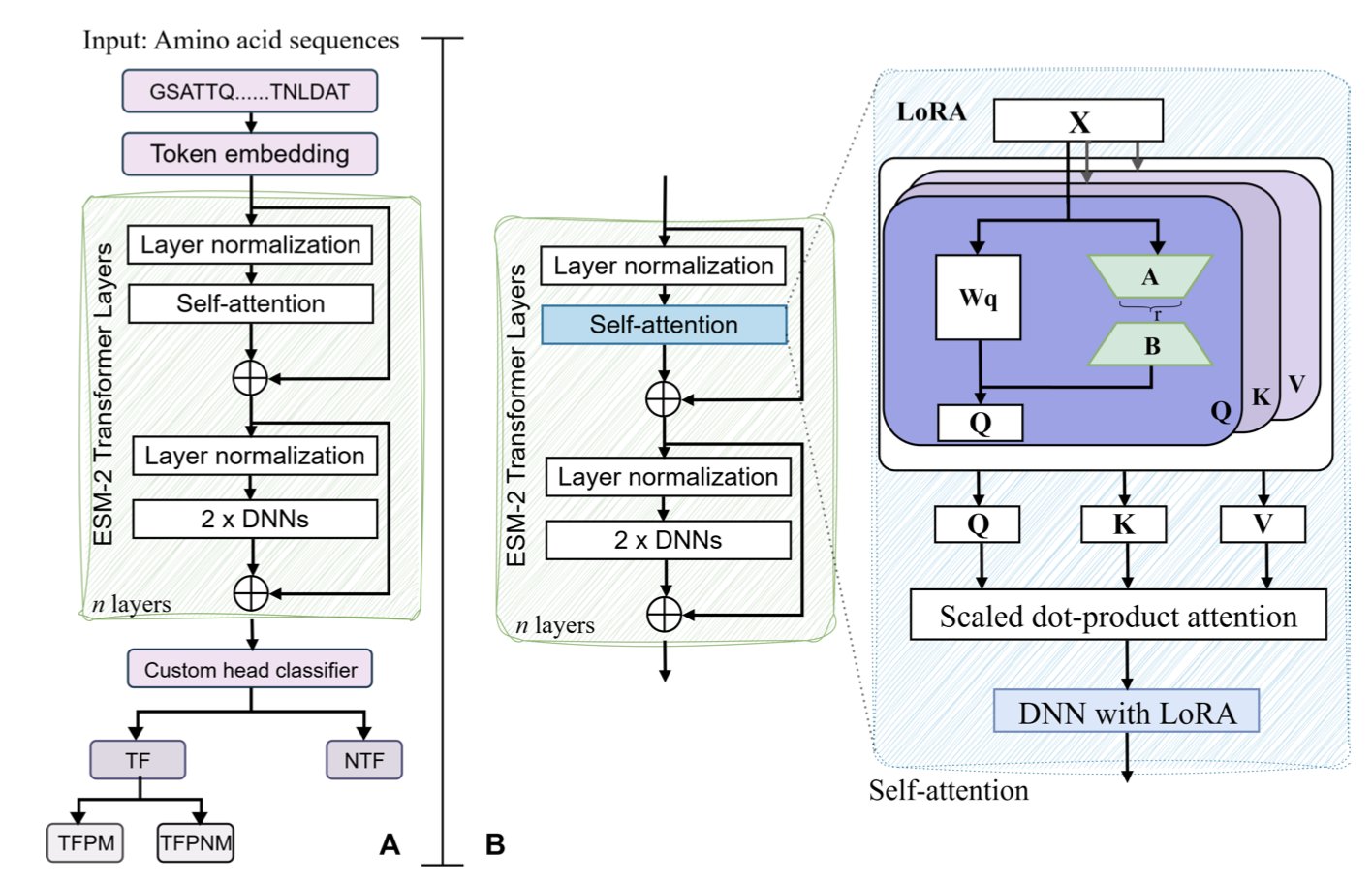
Protein language models (pLMs) are evolving from general-purpose tools into specialized experts. A new study on bioRxiv uses Meta’s ESM2 model and fine-tuning to tackle a core problem: identifying transcription factors (TFs) and their sensitivity to DNA methylation.
A Two-Layer Prediction Architecture
Transcription factors are numerous and have diverse functions. The study designed a two-layer prediction framework:
The first layer identifies the protein. The model analyzes a protein sequence to determine if it is a transcription factor. Compared to traditional sequence alignment, this method understands the “grammar” behind the sequence and is more accurate.
The second layer determines binding preference. After confirming a protein is a TF, the model analyzes whether it tends to bind to methylated DNA. This is a critical feature in epigenetics and cancer research.
The Balance of ESM2-650M and LoRA
Full fine-tuning is computationally expensive. After comparing models of different sizes, the authors found that ESM2-650M offered the best balance between parameter size and representation power.
Combined with LoRA (Low-Rank Adaptation), the study achieved parameter-efficient fine-tuning (PEFT). This strategy only requires training a small number of parameters, maintaining accuracy comparable to full fine-tuning while significantly lowering the computational cost. This makes it possible to deploy on local servers.
Explainability and Validation
To verify that the model truly understands the biological mechanisms, the authors extracted and visualized its attention weights.
The results showed that the regions the model paid high attention to matched known protein sequence motifs. The model accurately located the DNA-binding domains and the positions that determine methylation preference. This proves its predictions are based on biological principles, not just statistical noise.
📜Title: Fine-Tuning Protein Language Models Enhances the Identification and Interpretation of the Transcription Factors 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.11.27.691010v1