
Table of Contents
- Researchers developed scLDM, a new latent diffusion model that generates high-quality, scalable single-cell gene expression data, solving key problems with existing models.
- The ForceFM model brings physics-based force fields into deep learning. Its force-guided flow matching technique improves the accuracy and physical realism of protein-ligand docking predictions while lowering computational cost.
- HyperBind2 combines computational screening with experimental validation. It uses an iterative, multi-shot learning approach to improve the success rate and efficiency of computational antibody discovery.
- A new active learning method trains a separate model for each target and intelligently allocates computational resources, finding better compounds more efficiently in multi-target drug optimization.
- Proteins are dynamic. Apo2Mol uses a diffusion model to generate a ligand and adjust the pocket conformation at the same time, overcoming the challenge of designing drugs based on static apo structures.
1. scLDM: Generating Single-Cell Gene Expression Profiles with a Latent Diffusion Model
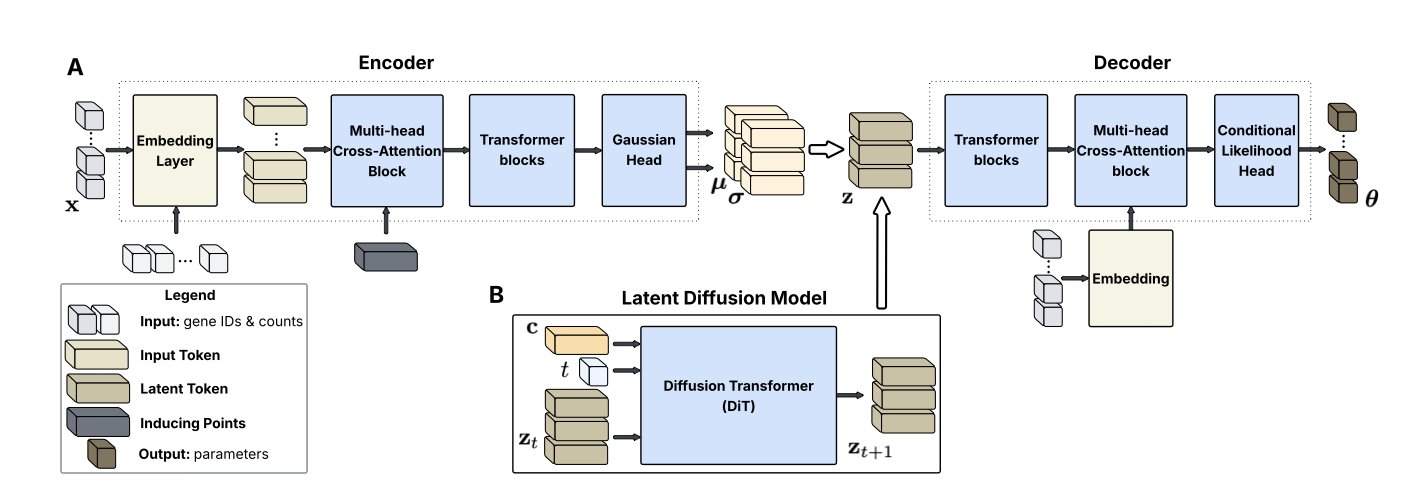
- New Architecture: scLDM uses a full Transformer architecture and a latent diffusion framework. Its Multi-head Cross-Attention Block (MCAB) solves issues with artificial gene ordering and training instability.
- Better Performance: On multiple benchmark datasets, scLDM reconstructs and generates data better than existing methods like scVI and CFGen.
- Controllable Generation and Application: The model supports conditional generation based on various attributes. It can simulate cellular responses to perturbations, and its learned embeddings perform well in downstream classification tasks.
Simulating high-quality single-cell gene expression data is a challenge in the field. Existing generative models often require a manually set gene order, which lacks a biological basis and makes training unstable. A new paper introduces the scLDM model to address these issues.
The scLDM model combines a Variational Autoencoder (VAE) and a Latent Diffusion Model.
First, the VAE compresses high-dimensional, complex single-cell gene expression data into a low-dimensional, compact “latent space.” To handle the “order-agnostic” nature of single-cell gene data, the researchers designed a Multi-head Cross-Attention Block (MCAB). During encoding, this block gathers information from all genes without bias. During decoding, it accurately restores information to each gene, respecting the data’s inherent biology.
Second, a diffusion model is trained in this information-dense latent space. scLDM uses a Diffusion Transformer to learn the complex structure of the latent space, rather than assuming a simple Gaussian distribution like a traditional VAE. The model can precisely control the generation process using “classifier-free guidance.” For example, it can be instructed to generate the gene expression profile for a “specific cell type” after being “stimulated by a certain drug,” and it can even handle multiple conditions at once. This is important for simulating complex biological scenarios, like how cells respond to genetic or chemical perturbations.
In comparisons with mainstream models like scVI and CFGen on a series of public datasets, scLDM showed higher Pearson correlation coefficients and lower reconstruction errors in data reconstruction and generation tasks. The data it generated was more realistic.
scLDM is also an effective feature extractor. The cell embedding representations it learns during data compression are useful for downstream biological discovery. For example, when analyzing a COVID-19 infection dataset, these embeddings effectively identified infected cells. When processing the large Tabula Sapiens 2.0 cell atlas, they also accurately classified cell types. This shows the model captures key biological information about cell states.
This work provides a solid and scalable framework for generative modeling of single-cell data. This kind of technology, which can accurately simulate how cells change under different perturbations, offers a powerful computational tool for understanding disease mechanisms and discovering new targets in drug development.
📜Title: Scalable Single-Cell Gene Expression Generation with Latent Diffusion Models 🌐Paper: https://arxiv.org/abs/2511.02986 💻Code: https://github.com/czi-ai/scLDM
2. A New Paradigm in AI Molecular Docking: The Force-Guided Flow Matching Model, ForceFM

Molecular Docking aims to find the most stable binding pose for a small molecule ligand inside a protein’s pocket. Traditional methods rely on a Scoring Function to evaluate conformations, while deep learning models have become a recent research focus. But many AI models lack physical logic when generating conformations, and their results may not follow the principle of minimum energy.
The ForceFM model introduces a “force-guided network,” combining the concept of physical force fields with a Flow Matching generative model. This is like installing a navigation system for a small ball trying to find the lowest point in a complex valley. The navigation system (the force-guided network) constantly indicates the direction of fastest energy drop, guiding the ball efficiently toward the minimum energy point—the most stable binding conformation—and avoiding random exploration.
The advantage of this method is accuracy. When tested on the PDBBind standard dataset, the conformations predicted by ForceFM had a lower root-mean-square deviation (RMSD) from the experimental structures, meaning its predictions are closer to the correct structure.
The second advantage is efficiency. Guided by the physical force field, the model makes fewer ineffective attempts, improving sampling efficiency. It converges to the ideal result in fewer computational steps, solving the high computational cost problem of many deep learning docking methods.
ForceFM is also highly adaptable. Studies show that the model adapts well when the force-guided network is trained with different scoring functions (like Vina, Glide, or Gnina), performing well in various blind docking and cross-domain evaluations. This indicates its core framework is generalizable and can be used as a plug-in to enhance different types of docking tools.
The model currently focuses on rigid docking, assuming the protein pocket’s shape is fixed. The real binding process involves interaction and dynamic conformational changes between the protein and the ligand, which is the next challenge for ForceFM to solve. Future research will involve integrating techniques that account for protein flexibility, such as Molecular Dynamics, to make the model more true to the biological process.
📜Title: ForceFM: Enhancing Protein-Ligand Predictions through Force-Guided Flow Matching 🌐Paper: https://openreview.net/pdf/ae92f0c9f07e2cab91eda336c624dcc34cb3fd7b.pdf
3. HyperBind2: Multi-Shot Learning to Improve Antibody Discovery Efficiency
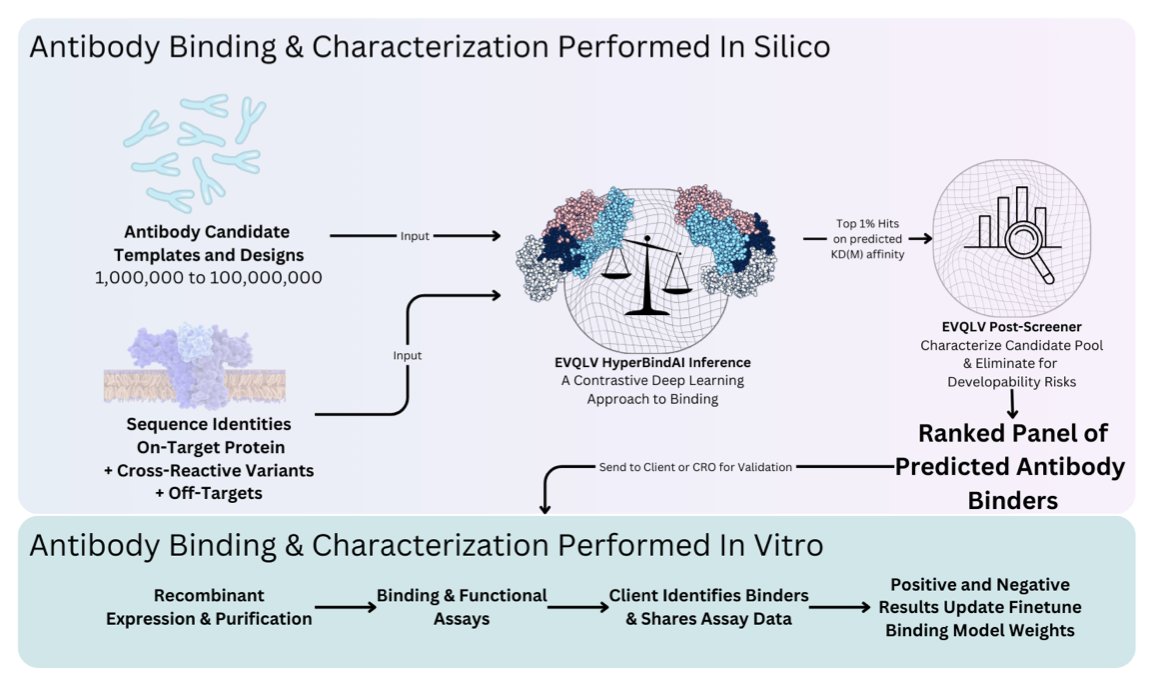
Antibody discovery is like finding a needle in a haystack. Traditional methods like display technologies or high-throughput screening are time-consuming, labor-intensive, and expensive. Computational methods hold promise, but their prediction accuracy is often not ideal, leading to many computationally screened candidates failing in experiments.
HyperBind2 offers a new approach. Instead of trying to find the perfect antibody in one shot, it uses a multi-shot learning strategy.
Here’s how it works:
First, the platform performs a large-scale computational pre-screening based only on the target’s amino acid sequence. The model embeds the antibody and antigen sequence information into a shared representation space and predicts binding affinity by learning their geometric relationships. The advantage here is that it doesn’t require the target’s 3D structure, which is one of the most time-consuming parts of drug development.
Next, a small number of candidates are selected from the vast pool of computational hits for experimental validation. This stage is small-scale, with the goal of collecting experimental data—confirming which molecules are effective and which are not.
The most critical step is feeding this new experimental data back into the model for retraining and fine-tuning. By learning from real binding data, the model can more accurately understand the binding preferences of a specific target, improving its performance in the next round of predictions.
Through this cycle of “computational prediction -> experimental validation -> model optimization,” HyperBind2’s predictive power iteratively improves. In a project targeting a difficult multi-pass transmembrane receptor, the experimental success rate reached 21% after just three rounds of optimization. In the computational field, especially for such tricky targets, this number is a huge leap. It means researchers can significantly reduce the number of molecules they need to validate in wet labs, saving time and resources.
Future Applications
HyperBind2 supports various antibody formats, including single-chain antibodies (scFvs), nanobodies (VHHs), and full-length IgGs. This allows it to be integrated into the development pipelines for advanced therapies like Chimeric Antigen Receptor T-cell (CAR-T) therapy or Bispecific T-cell Engagers (BiTEs).
The platform is now open-source for academic use and available as a commercial version for industry. This allows labs without a computational background to directly obtain optimized antibody sequences for their subsequent experimental development.
📜Paper Title: HyperBind2: Multi-Shot Learning Enables Progressive Improvement in Computational Antibody Discovery 🌐Paper Link: https://www.biorxiv.org/content/10.1101/2025.11.06.687005v2
4. A New AI Drug Discovery Strategy: Divide and Conquer
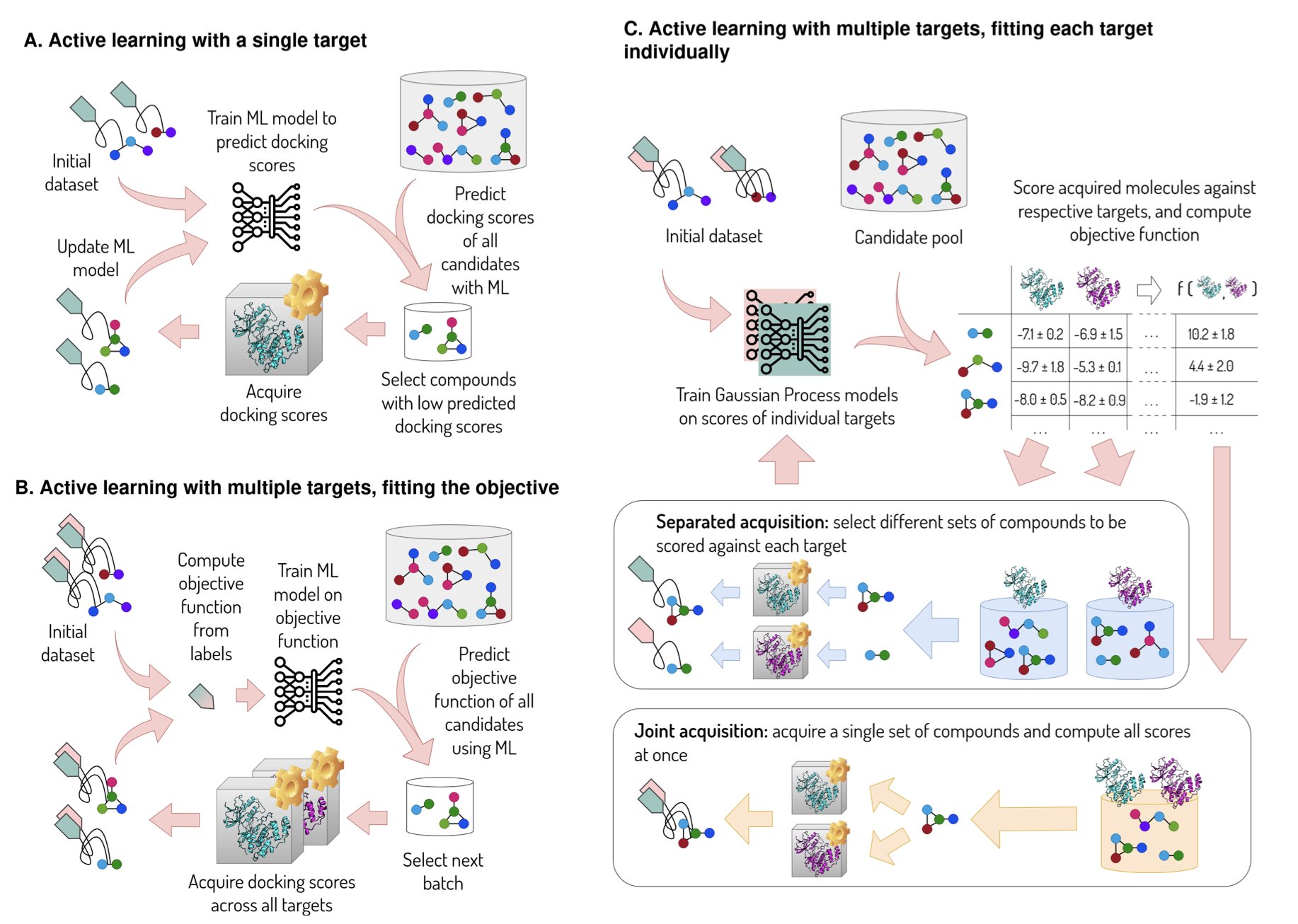
Drug development is a balancing act. We want a compound to be potent against its intended target while avoiding all off-targets. When there is more than one property to optimize—for example, balancing activity at target A, selectivity over target B, and solubility—the problem gets complicated.
Traditional computational methods usually bundle all objectives into a single “objective function” and have one machine learning model optimize this total score.
Splitting Tasks Works Better
The authors of this paper took a different approach, using a “separated acquisition” strategy. They assembled a “team of experts,” with each model responsible for one task.
Here’s how it works: 1. A dedicated machine learning model is trained for each property to be optimized. For instance, one model predicts a molecule’s binding affinity to JAK2, while another predicts its binding to other kinases. 2. In each round of active learning, the algorithm evaluates where to spend its computational resources next to maximize the final total score. Should it calculate scores for a few more molecules on JAK2, or on the off-targets?
This decision-making process is intelligent. If a property has a large impact on the total score, or if a model’s predictions are highly uncertain and have room for improvement, the algorithm allocates more of its computational budget (like expensive molecular docking or dynamics simulations) to it. This is like a project manager investing funds first in the most critical or weakest parts of a project.
Speaking with Real Data
The researchers used the public DOCKSTRING dataset for a retrospective validation, simulating two drug discovery scenarios:
- Pursuing selectivity: Optimizing a JAK2 inhibitor to have high activity against JAK2 while avoiding structurally similar kinases.
- Pursuing broad-spectrum activity: Optimizing a PPAR agonist to activate multiple PPAR subtypes simultaneously.

The results showed that in both scenarios, the “separated” strategy found high-quality compounds at a higher rate than the traditional “joint” strategy. With the same amount of computational resources, this new method discovered promising molecules faster.
Practical Value and Outlook
This method has several advantages.
First, it aligns with the intuition of medicinal chemists. In real projects, researchers often focus on specific problems, like solving for activity first and then dealing with selectivity. This “divide and conquer” computational strategy simulates and optimizes a real-world development process.
Second, it’s highly scalable. Real drug discovery involves more than just two or three properties; it also includes ADME (absorption, distribution, metabolism, excretion), toxicity, and more. This framework can easily accommodate more “expert models” to handle more complex optimization tasks.
The authors also noted the method’s limitations. For example, it requires a clearly defined objective function upfront. Also, the method does not currently explicitly account for molecular diversity, which could limit the search to a narrow chemical space.
Their next plan is to apply this method to free energy calculations. Molecular docking scores are cheap to compute but rough, whereas free energy calculations based on molecular dynamics (MD) are more accurate but expensive. Using this smart allocation strategy to decide which molecules are worth the costly free energy calculations could improve efficiency in the lead optimization stage.
📜Paper: A Simple Compound Prioritization Method for Drug Discovery Considering Multi-Target Binding 🌐Paper Link: https://doi.org/10.26434/chemrxiv-2025-vqg5k 💻Code Link: https://github.com/MobleyLab/active-learning-notebooks/blob/main/MultiobjectiveAL.ipynb
5. Apo2Mol: A New Path for AI Drug Design that Tackles Protein Flexibility
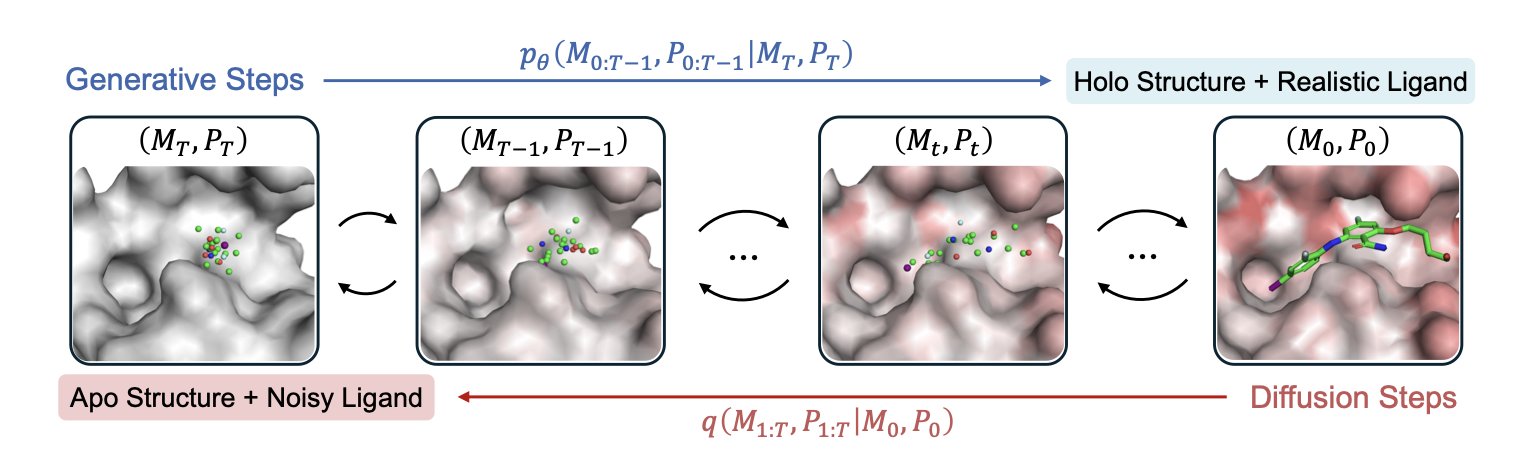
In medicinal chemistry, we must always remember one thing: proteins are not static rocks.
Traditional structure-based drug design (SBDD) often faces a difficult situation: you only have the protein’s unbound Apo structure. It’s like designing a glove for an open hand when all you have is a photo of a fist.
When a drug molecule enters a protein pocket, it induces a conformational change, a process known as “induced fit.” Most current AI models assume the protein is rigid. This leads to molecules that look perfect in theory but have poor binding affinity in reality.
Apo2Mol solves this problem with a Dynamic Pocket-Aware Diffusion Model. This model generates the drug molecule while simultaneously adjusting the protein pocket’s shape in real time. It’s like kneading dough, allowing the molecule and the pocket to adapt to each other until they reach the lowest-energy bound state (the Holo state).
The team performed a deep cleaning of the Protein Data Bank (PDB) to curate a dataset of over 24,000 experimentally validated Apo-Holo structure pairs. The model used this data to learn the real physical rules of binding—how residues rotate and side chains move out of the way when a ligand enters—allowing it to accurately predict new ligand-protein complexes.
Apo2Mol excels at generating high-affinity ligands and producing realistic pocket conformations. This is valuable for developing first-in-class drugs, where often only the Apo structure is known and the bound conformation is a mystery. For researchers in computer-aided drug design (CADD) who are tackling highly flexible or hard-to-drug targets, this is a breakthrough approach.
📜Title: Apo2Mol: 3D Molecule Generation via Dynamic Pocket-Aware Diffusion Model 🌐Paper: https://arxiv.org/abs/2511.14559v1