
Table of Contents
- The DualMPNN model uses structural information from similar proteins to more accurately design amino acid sequences that can fold into a specific 3D structure.
- The SpecLig framework combines a hierarchical graph neural network with an energy-guided diffusion model to improve the target specificity of de novo ligand design.
- By systematically evaluating 29 models, NABench reveals performance differences among various nucleotide large language models in predicting sequence function, pointing the way for future optimization.
- The GraphCliff model uses an innovative gating mechanism to simultaneously capture a molecule’s local details and global information, improving prediction accuracy for activity cliff compounds.
- This specialized graph neural network architecture solves the trade-off between computational efficiency and accuracy in macromolecule pairwise similarity prediction, making it a useful tool for large-scale drug screening.
1. Dual-Stream MPNN: Using Structural Alignments for Inverse Protein Folding
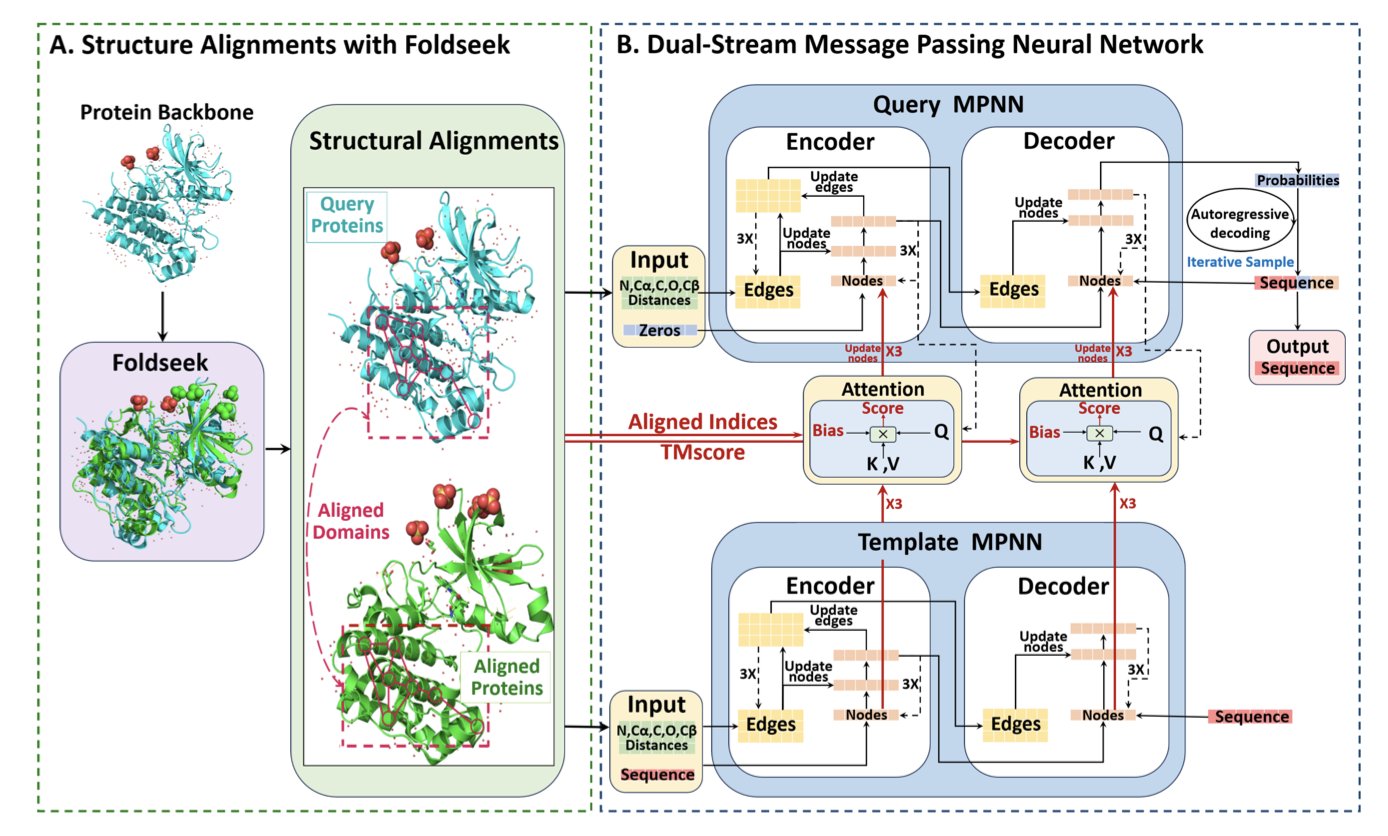
A core challenge in protein design is inverse protein folding: given a specific 3D structure, how do you find the amino acid sequence that will fold into it? It’s like looking at a building’s blueprint and figuring out the exact type and order of bricks needed. DualMPNN offers a new way to solve this.
When designing a new chair, you might look at how existing chairs are built. DualMPNN works similarly. It doesn’t just analyze the target 3D structure (the query structure); it also finds structurally similar proteins from a database (structural homologs). This is like referencing classic designs while creating a new one.
At the heart of the DualMPNN model is a dual-stream information processing system. One stream analyzes the geometric features of the target structure itself. The other stream studies the sequence and structural patterns of its homologs. The model then allows these two streams of information to interact. For example, rules about amino acid arrangements learned from the reference structures can help guide the choice of amino acids at key positions in the target structure.
This approach works well. Researchers tested DualMPNN on industry-standard benchmarks like CATH, TS50, and T500. The sequence recovery rates were 65.51%, 70.99%, and 70.37%, respectively. This is an improvement of at least 12 percentage points over the high-performing ProteinMPNN model. The improvement means the designed sequences are more likely to successfully fold into the intended structure.
To check if the designed sequences were biologically viable, the researchers used AlphaFold2 and AlphaFold3 to predict their folded structures. Most sequences folded back to the target structure with high accuracy, showing that the proteins designed by DualMPNN are reliable.
Using structural information from known similar proteins can simplify the design of new proteins and increase the success rate. This opens up new possibilities for designing proteins with specific functions from scratch, especially in cases where sequence data is limited.
📜Title: DualMPNN: Harnessing Structural Alignments for High-Recovery Inverse Protein Folding 🌐Paper: https://openreview.net/pdf/faba58f19ead837d5a34f3a9aef572473bfd2447.pdf
2. SpecLig: Energy-Guided AI Ligand Design for Better Target Specificity
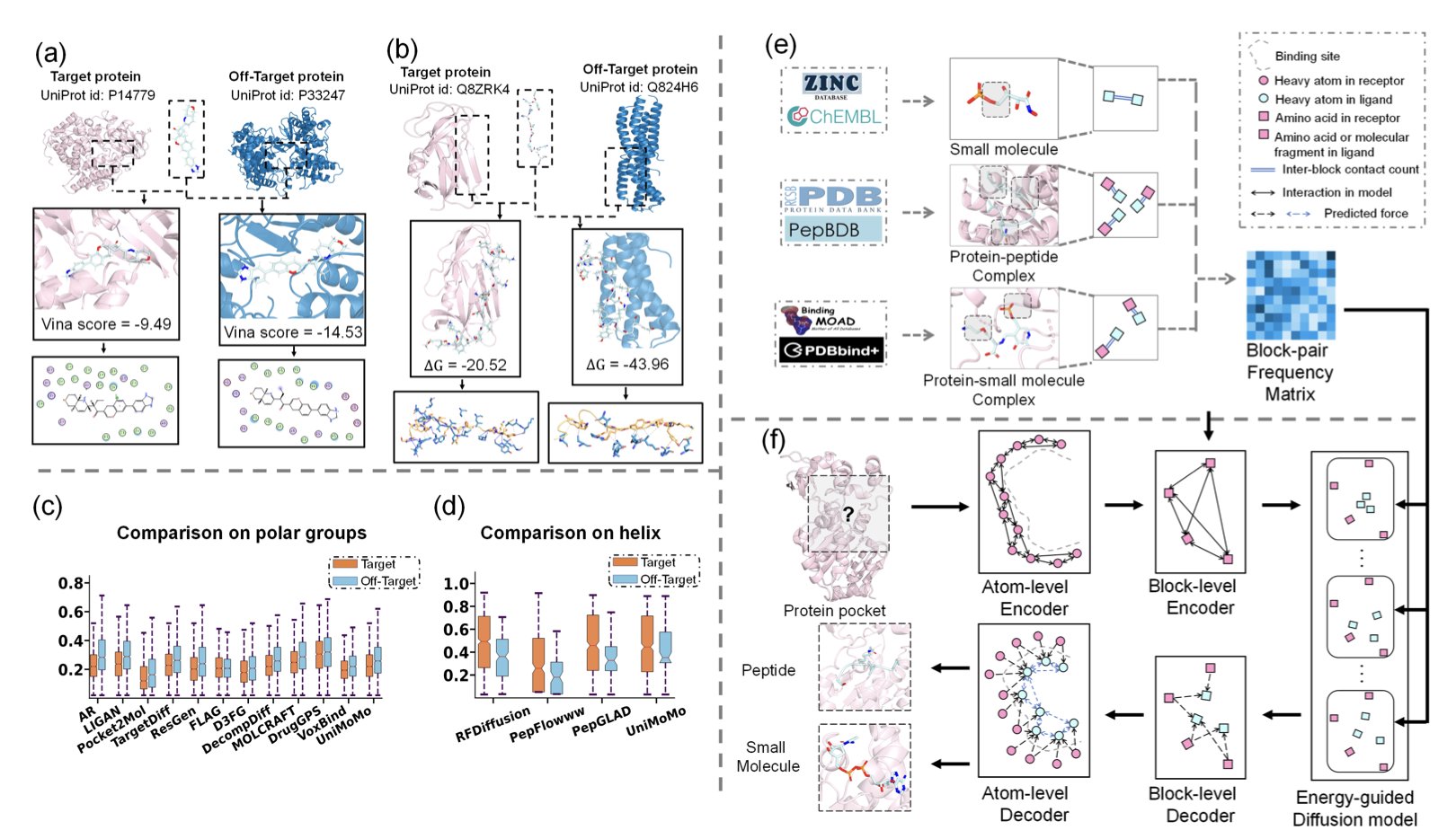
Drug discovery has a constant goal: make drug molecules that bind precisely to their intended target while avoiding others. It’s like designing a key that only opens the right lock. Off-target effects can cause side effects or even serious toxicity.
A new computational framework called SpecLig brings a new approach to this specificity problem.
Traditional drug design methods focus on improving a molecule’s binding affinity to its target. SpecLig goes a step further by also working to ensure the molecule is tailored for that specific target, achieving specificity.
To do this, the researchers built a hierarchical model that avoids the noise introduced by working directly at the atomic level. They treat proteins and ligands as graph structures made of different “functional blocks.” This approach better captures the local chemical environment and overall topology of the molecules, preserving meaningful chemical fragment information.
The core of SpecLig is a hierarchical variational autoencoder (VAE) and an energy-guided diffusion model. The model first learns which blocks are more likely to form stable interactions by studying a large dataset of natural protein-ligand complexes.
When generating a new molecule, the energy-guided diffusion model uses these statistical “energy preferences” to guide its construction. It favors generating conformations that have stronger geometric and chemical complementarity with the target pocket, which in turn improves specificity.
Researchers tested SpecLig on standard small-molecule and peptide datasets. The results showed that ligands generated by SpecLig had significantly higher specificity scores while maintaining comparable affinity to those from existing methods. Case studies also demonstrated that the method can effectively optimize a molecule’s pocket specificity and reduce off-target risks.
Designing small-molecule drugs remains a big challenge. The complexity and sensitivity of chemical space mean there is still a long way to go. The researchers acknowledge that future versions could integrate more physicochemical information to further improve the model’s performance.
SpecLig provides a powerful new tool. It demonstrates that by using clever modeling and data, specificity can be considered early in the drug design process, paving the way for safer and more effective drug candidates.
📜Name: SpecLig: Energy-Guided Hierarchical Model for Target-Specific 3D Ligand Design 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.11.06.687093v2
3. NABench: A Standardized Test for Large Nucleotide Models
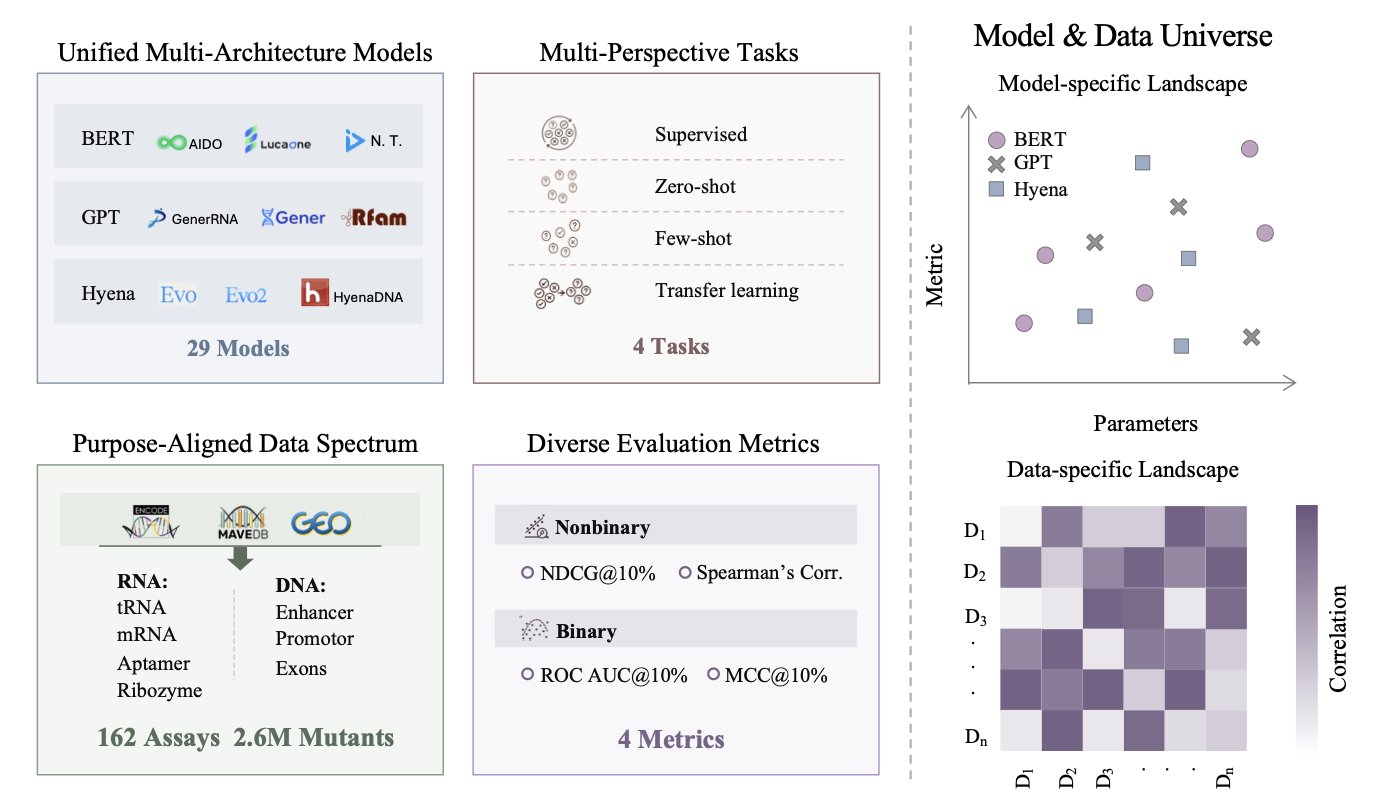
In protein engineering, using benchmarks to evaluate the performance of protein Large Language Models (LLMs) is standard practice. But for nucleic acids like DNA and RNA, a comprehensive, unified evaluation standard has been missing. NABench fills that gap.
Understanding how a gene mutation affects its function is critical for drug development. A tiny change in a DNA sequence can cause a protein to malfunction and lead to disease. Accurately predicting the consequences of these changes can help in designing targeted drugs or developing gene therapies. NABench was created to systematically assess the ability of existing Nucleotide Foundation Models (NFMs) to predict these functional impacts.
The researchers collected and organized data from 162 high-throughput screening experiments. The data covers a wide range of DNA and RNA families, including promoters, enhancers, ribozymes, and RNA aptamers, and includes 2.6 million mutated sequences with functional labels. With a standardized dataset and evaluation metrics, different models can finally be compared on a level playing field.
The evaluation included 29 major nucleotide models and was comprehensive, covering four scenarios: zero-shot, few-shot, supervised, and transfer learning.
In the zero-shot scenario, where models make predictions based only on what they’ve learned from vast amounts of sequence data, state-space models like Evo performed the best. This suggests these types of models learn more robust knowledge from evolutionary information and have better generalization ability.
In the supervised learning scenario, where models are fine-tuned for a specific task, BERT-like models had an advantage. BERT-like architectures are good at incorporating task-specific data, and fine-tuning allows them to adapt better to the problem at hand.
The researchers also introduced the SELEX dataset to test the models’ generalization ability. SELEX is an in-vitro screening technique that produces large numbers of synthetic sequences not found in nature. They found that models trained on natural sequences performed poorly when predicting the function of these artificial sequences. This exposes a core weakness of current models: they have learned the rules of natural evolution but lack the ability to explore the vast space of non-natural sequences.
NABench provides a yardstick for the field of nucleic acid sequence research. It highlights the strengths and weaknesses of existing models and points the way forward for computational drug discovery and synthetic biology: developing next-generation nucleotide models that can both understand the rules of natural evolution and explore artificially designed spaces.
📜Title: NABench: Large-Scale Benchmarks of Nucleotide Foundation Models for Fitness Prediction 🌐Paper: https://arxiv.org/abs/2511.02888
4. GraphCliff: A New Graph Neural Network for Predicting Activity Cliffs
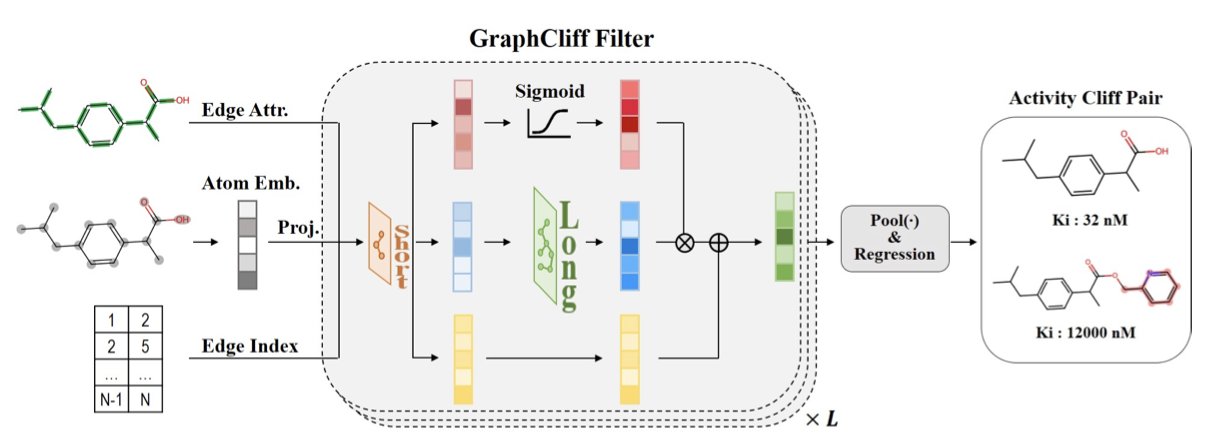
A tricky problem in drug discovery is “activity cliffs.” Two molecules might differ by just one atom or functional group, but their biological activity can be completely different. Traditional Quantitative Structure-Activity Relationship (QSAR) models often fail to capture these dramatic shifts in activity caused by such small structural changes.
To address this challenge, researchers developed a Graph Neural Network (GNN) model called GraphCliff.
The model introduces a new gating mechanism, which acts like an information-regulating valve. It helps the model pay attention to two types of information at the same time: a molecule’s local details, like specific chemical bonds or functional groups (short-range information), and its overall structural features (long-range information).
When traditional GNN models process molecular graphs, information becomes more and more uniform after several rounds of message passing. This leads to “over-smoothing,” which can obscure key local features. GraphCliff’s gating mechanism balances local and global information, allowing the model to recognize local substructures while also understanding the molecule’s overall characteristics.
Comparative experiments on the MoleculeACE benchmark dataset showed that GraphCliff performed better than existing models at predicting the activity of both ordinary compounds and, especially, activity cliff compounds. This confirmed its effectiveness at capturing subtle structural differences.
A deeper analysis showed that GraphCliff’s gating mechanism played a key role. It allowed the model to better retain long-range information and ensured that the features of different atoms in the molecule remained distinct during computation. Visualizations also revealed that the model could accurately focus its attention on the key substructures that cause activity cliffs. This not only improves prediction accuracy but also gives us a better chemical understanding of the model’s decisions.
📜Paper: GraphCliff: Short-Long Range Gating for Subtle Differences but Critical Changes 🌐Link: https://arxiv.org/abs/2511.03170
5. A New GNN for Efficient and Accurate Macromolecule Similarity Prediction
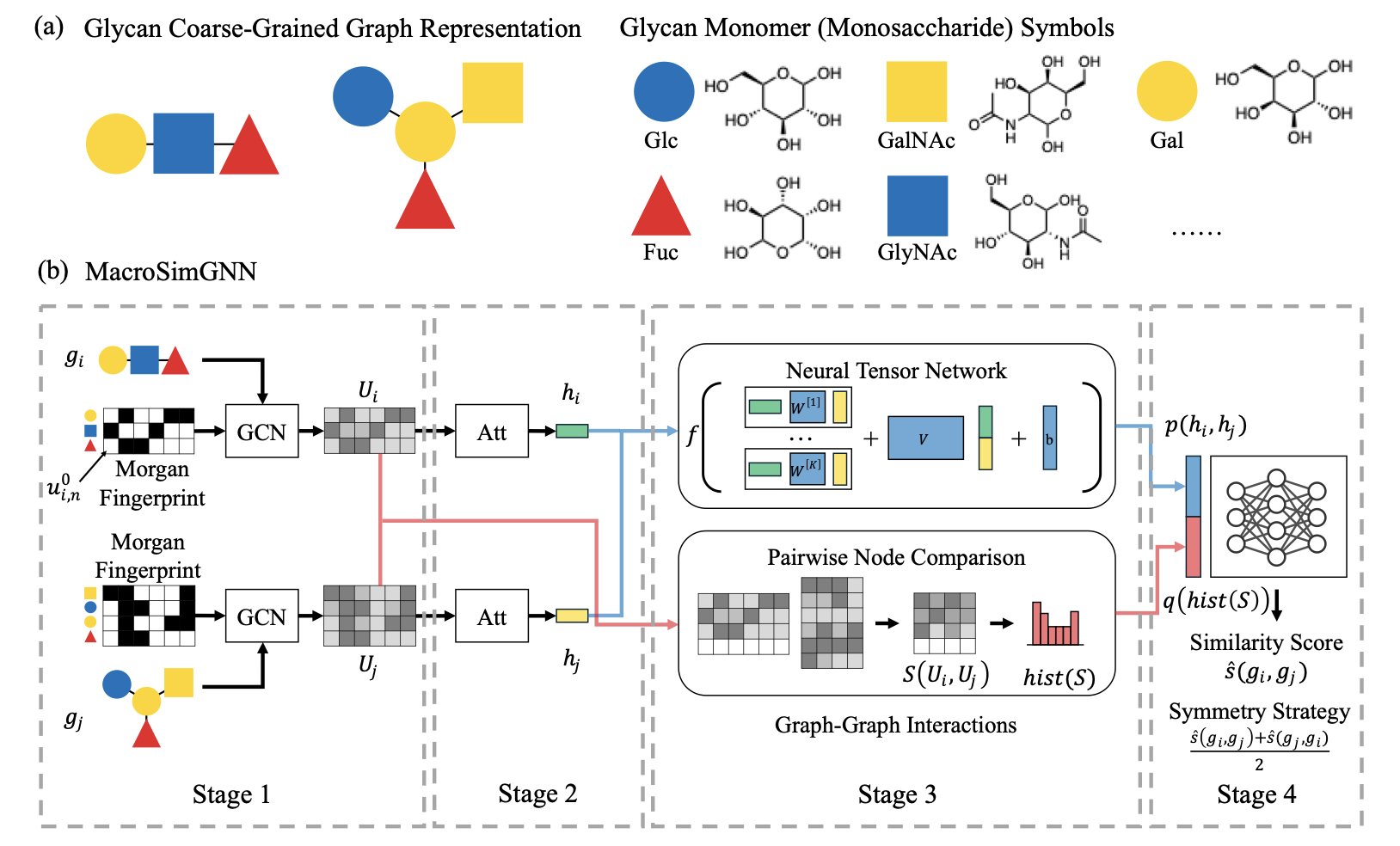
Computer-Aided Drug Design (CADD) is quite mature for small molecules, but it still faces challenges in the world of macromolecules. Quickly and accurately predicting the pairwise similarity of large molecules has always been a difficult problem. The authors of this paper have introduced a Graph Neural Network (GNN) to tackle it.
The authors designed a specialized architecture adapted for macromolecule data. Since the function of a macromolecule is determined by its 3D structure, the model uses these structural properties to capture the complex internal interactions and relationships. It’s like looking past a machine’s exterior to understand how the gears inside mesh together.
In rigorous testing across different datasets, this method performed very well. Compared to traditional alignment methods, the new model’s prediction accuracy was much higher, and it was robust, remaining stable even with small fluctuations in the data.
Computational efficiency is key for industrial applications. As biological data grows exponentially, an algorithm’s speed determines its practical value. This method can handle large-scale macromolecule data. In the early screening stages of drug discovery and protein engineering, it can help researchers quickly process huge numbers of samples and precisely identify key structural analogs.
📜Title: Macro-sim-gnn-efficient-and-accurate-prediction-of-macromolecule-pairwise-similarity-via-graph-neural-network 🌐Paper: https://doi.org/10.26434/chemrxiv-2024-8hs2k-v2