
Table of Contents
- MedRule-KG embeds the “hard logic” of knowledge graphs into the LLM generation process, acting like a constraint to significantly reduce basic scientific errors in drug design.
- MEHC-Curation uses a standardized validation, cleaning, and normalization process to solve the “garbage in, garbage out” problem in drug discovery data, improving the predictive power of QSAR models.
- The GRASP framework uses a clever prompt engineering method to enable Large Language Models to efficiently and accurately infer complex gene relationships, even across species.
- This study uses explainable AI (SHAP) to show that deep learning models genuinely understand the physicochemical mechanisms behind peptide retention time and fragmentation patterns.
- ANUBI packages complex molecular dynamics simulations and free energy calculations into an automated tool, allowing non-computational experts to quickly screen and optimize protein or peptide drug candidates.
1. MedRule-KG: Adding “Logic Guardrails” to AI Drug Discovery
Drug discovery researchers know the common problem with Large Language Models (LLMs): they can write beautiful poetry, but they often spout nonsense with a straight face when predicting reaction products or designing molecules. In chemistry and biology, these “hallucinations” are a deal-breaker. A carbon atom can’t have five bonds, no matter how elegant the AI’s proposed structure is.
MedRule-KG tries to fix this. Instead of endlessly retraining the model with more data, it fits the model with an “exoskeleton.”
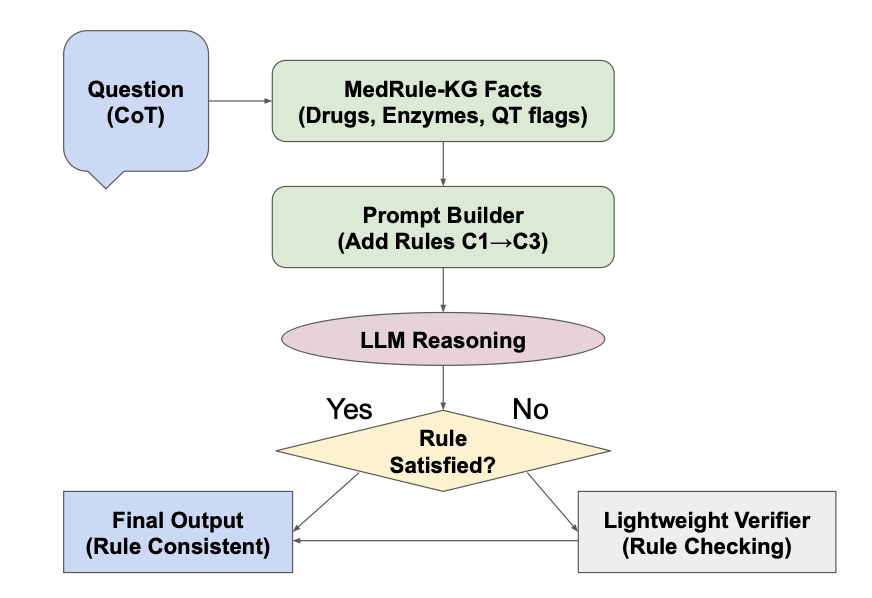
How It Works
Think of it like setting rules for a brilliant student who has a habit of making things up.
First, it introduces a lightweight knowledge graph. This acts like a pocket textbook, containing verified biomedical facts.
The core component is the Soft Constraint Controller. MedRule-KG intervenes in the LLM’s probability-based generation process. If the next word would violate a rule in the knowledge graph (such as metabolic compatibility), the controller lowers its probability, steering the model back to a valid path. This maintains logical accuracy without sacrificing fluency.
Finally, a Deterministic Verifier acts as an “auditor.” It reviews the model’s output after generation and rejects anything that violates predefined biochemical rules.
Performance
The data is straightforward. On key tasks like reaction feasibility, metabolic compatibility, and toxicity screening, the system reduced rule violations by 83.2% compared to strong baseline models. This means we are more likely to get molecules that can actually be synthesized, not just theoretical curiosities.
Speed is also important. The entire process—prompting, generation, and verification—handles a single query in under 200 milliseconds. This makes it practical for high-throughput screening or interactive drug design.
Neuro-symbolic AI is a promising direction. Relying on neural network black boxes to simply “guess” chemical principles has its limits. Integrating explicit scientific rules (symbolic logic) is a path toward more reliable AI.
📜Title: MedRule-KG: A Knowledge-Graph–Steered Scaffold for Reliable Mathematical and Biomedical Reasoning 🌐Paper: https://arxiv.org/abs/2511.12963v1
2. MEHC-Curation: An Open-Source Framework for Cleaning QSAR Data
Researchers in Computer-Aided Drug Design (CADD) or QSAR modeling often face a common problem. Molecular data downloaded from public databases frequently contains salts, solvent molecules, invalid SMILES strings, and even conflicting activity data for the same compound. This “dirty data” leads directly to “garbage in, garbage out.”
The MEHC-Curation Python framework was built to solve this. It provides a complete, automated processing pipeline.
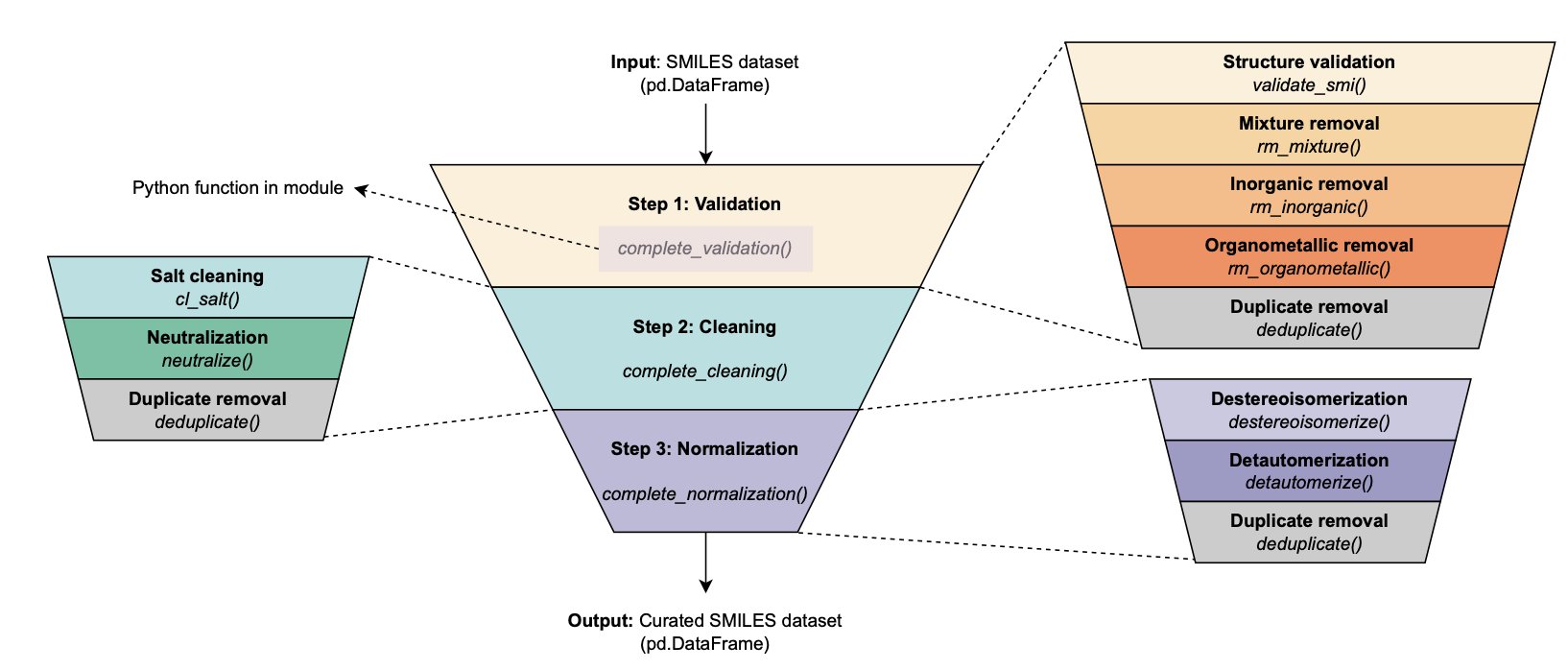
How It Works
MEHC-Curation breaks down data processing into three core steps:
- Validation: Identifies and removes invalid chemical structures.
- Cleaning: Strips away salts and solvent molecules, keeping only the core active pharmaceutical ingredient.
- Normalization: Standardizes tautomers and aromatic ring representations to ensure a single, canonical representation for each molecule.
The framework also has a built-in deduplication feature that handles duplicate molecules according to predefined rules, such as taking the average of their activity values.
Performance
The authors tested the framework on 15 benchmark datasets. Machine learning models trained on the curated data consistently outperformed models trained on the original, unprocessed data. This confirms the critical role of data quality in AI-driven drug discovery.
Advantages
The tool supports parallel processing to handle large datasets efficiently. Its traceability is especially useful: the system generates detailed logs that record every change. You can clearly see why a molecule was removed, whether due to a formatting error or an invalid structure.
It’s simple to use. All you need is a .csv file containing SMILES strings. This saves drug discovery researchers a lot of preprocessing time.
📜Title: MEHC-Curation: A Python Framework for High-Quality Molecular Dataset Curation 🌐Paper: https://doi.org/10.26434/chemrxiv-2025-3x7gq
3. GRASP: Using Prompt Engineering with Large Models to Infer Gene Networks
Inferring gene regulatory networks has always been a tough problem in bioinformatics. We have more and more data, but figuring out which genes regulate each other from this vast amount of information is still a huge challenge. Existing computational methods all have their limitations.
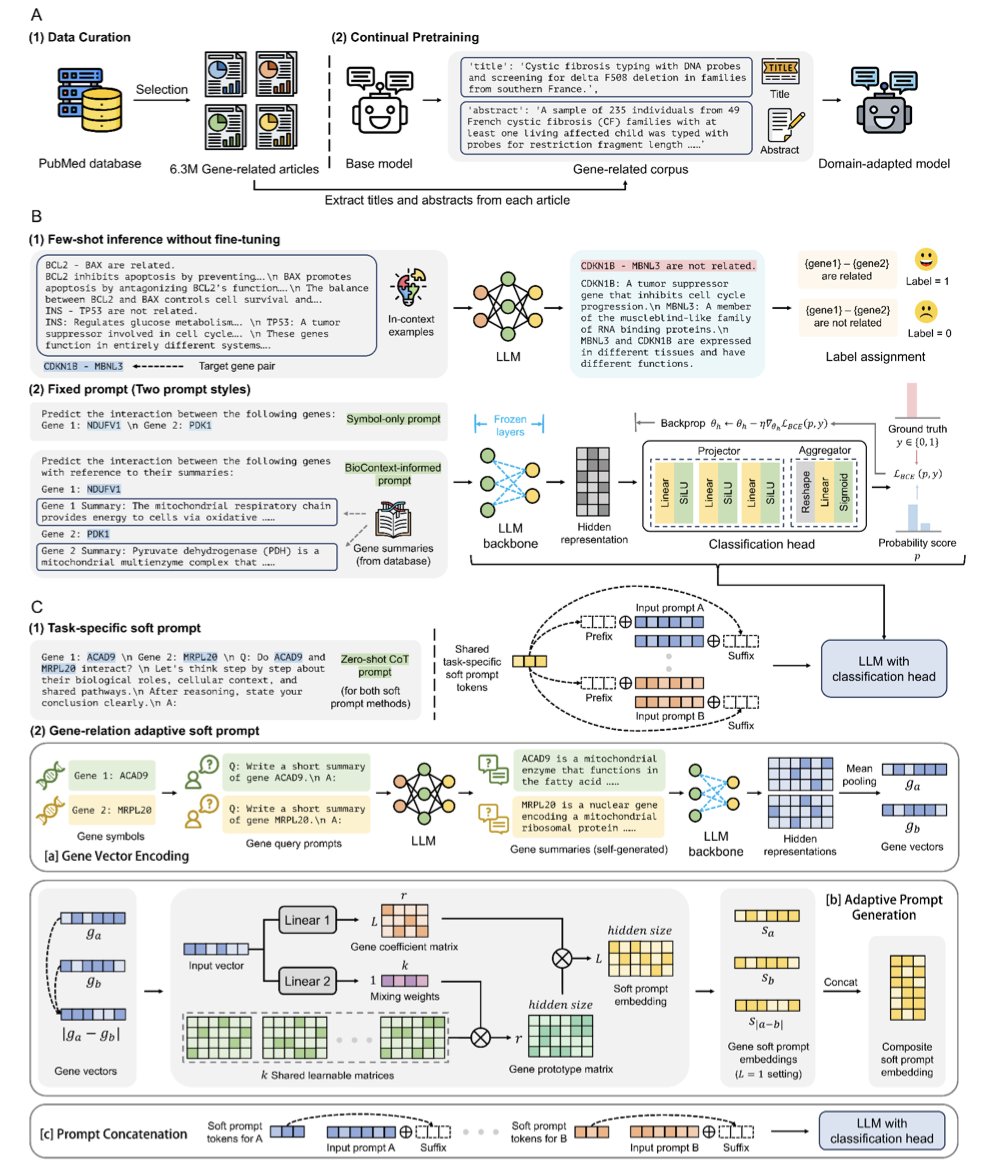
The GRASP (Gene-Relation Adaptive Soft Prompt) framework offers a new approach. Instead of retraining a model from scratch, it uses a parameter-efficient method to adapt existing Large Language Models (LLMs).
Here’s one way to think about it. A pre-trained LLM is like an expert with broad but not specialized knowledge. If you ask it directly about the relationship between two genes, its answer might be generic. GRASP’s method is to hand this expert a highly customized “cheat sheet”—an “adaptive soft prompt”—along with the question.
This cheat sheet is tailored for each pair of genes and contains their specific features and clues about their potential relationship. With this prompt, the expert (the LLM) can make a much more accurate judgment. During this process, the pre-trained model’s parameters remain frozen. Only the module that generates the “cheat sheet” needs to be trained. This approach leverages the LLM’s vast prior knowledge while remaining parameter-efficient.
In large-scale benchmarks for protein-protein interaction (PPI) prediction, GRASP outperformed all baseline methods, achieving a ROC-AUC score as high as 0.937.
The model is also generalizable. A GRASP model trained on human data can directly predict gene interactions in mice. This cross-species transferability has potential applications in drug development and translational medicine. The model also performs well in inferring other biological networks, like phosphorylation networks.
GRASP also shows potential for “error correction” and “discovery.” It can identify gene interactions that were previously mislabeled in databases. This means GRASP can not only predict unknown relationships but also review and correct existing knowledge bases. It can act as a discovery engine, helping us find new biological connections within massive datasets.
📜Title: GRASP: Gene-Relation Adaptive Soft Prompt for Universal Gene Network Inference 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.20.683485v1
4. Opening the Peptide AI Black Box: SHAP Reveals the Chemical Principles Behind Predictive Models
Deep learning models excel at processing proteomics data, accurately predicting properties like retention time, collision cross-section (CCS), and even fragment intensity. The main challenge has been the “black box” problem: we need to confirm that these models are making decisions based on correct physicochemical principles, not just fitting biases in the data.
This study uses SHAP (Shapley Additive Explanations) to dissect peptide prediction models. The results show that these models do, in fact, understand chemical principles.
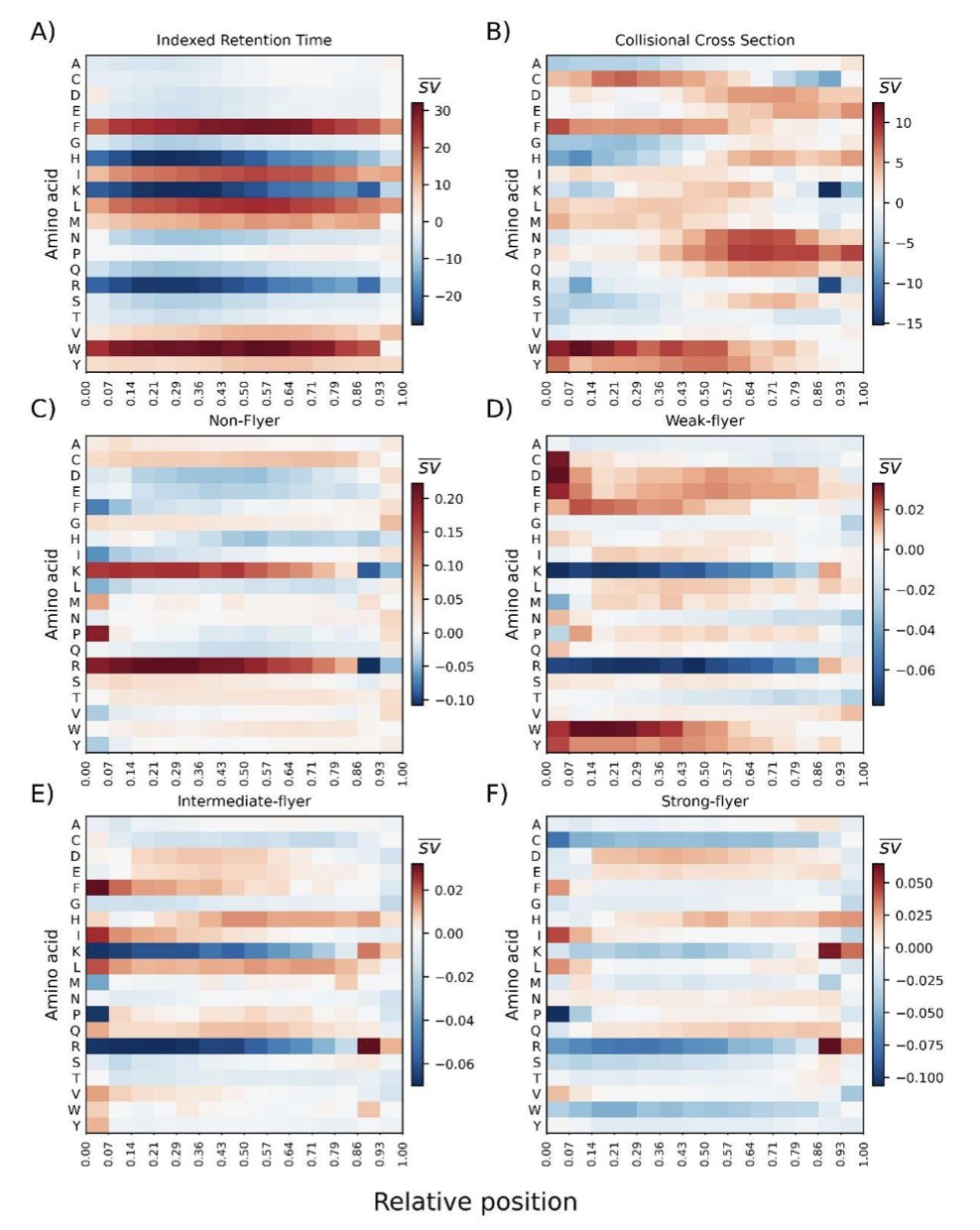
Retention Time: The Model Learned Hydrophobicity
An analysis of a liquid chromatography retention time predictor found that the model automatically assigned positive SHAP values (which increase retention time) to hydrophobic amino acids and negative values to hydrophilic ones.
The model learned this from sequence data alone, without ever being given the Kyte-Doolittle hydrophobicity scale. When its derived SHAP values were compared to classic amino acid indices, the Pearson correlation coefficient was 0.973. The AI essentially developed its own “physicochemical intuition,” one that closely matches what chemists have measured experimentally.
Fragmentation Mechanism: Recreating the Mobile Proton Model
An analysis of tandem mass spectrometry (MS/MS) fragmentation patterns showed that the model is highly sensitive to basic amino acids like arginine and lysine.
Peptide fragmentation follows the “mobile proton model,” where the ease of breaking a peptide bond depends on proton mobility. The SHAP analysis revealed that the model captured how strongly basic residues sequester protons, and it correctly predicted the resulting shifts in fragment intensity distribution. It also distinguished the subtle differences between charge-directed and charge-remote fragmentation pathways.
From Black Box to White Box
Experiments with custom-synthesized peptides further confirmed that the model is sensitive to mutations at specific sites. This demonstrates the model is simulating the chemical behavior of the molecule.
For scientists in drug discovery and proteomics, these models can be trusted. SHAP can be an important bridge connecting data science with complex reaction mechanisms, such as post-translational modifications (PTMs) or electron-transfer/capture dissociation (ETD/ECD).
📜Title: Systematic evaluation of peptide property predictors with explainable AI technique SHAP 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.11.19.689259v1
5. ANUBI: An Automated Platform for Optimizing the Affinity of Protein and Peptide Drugs
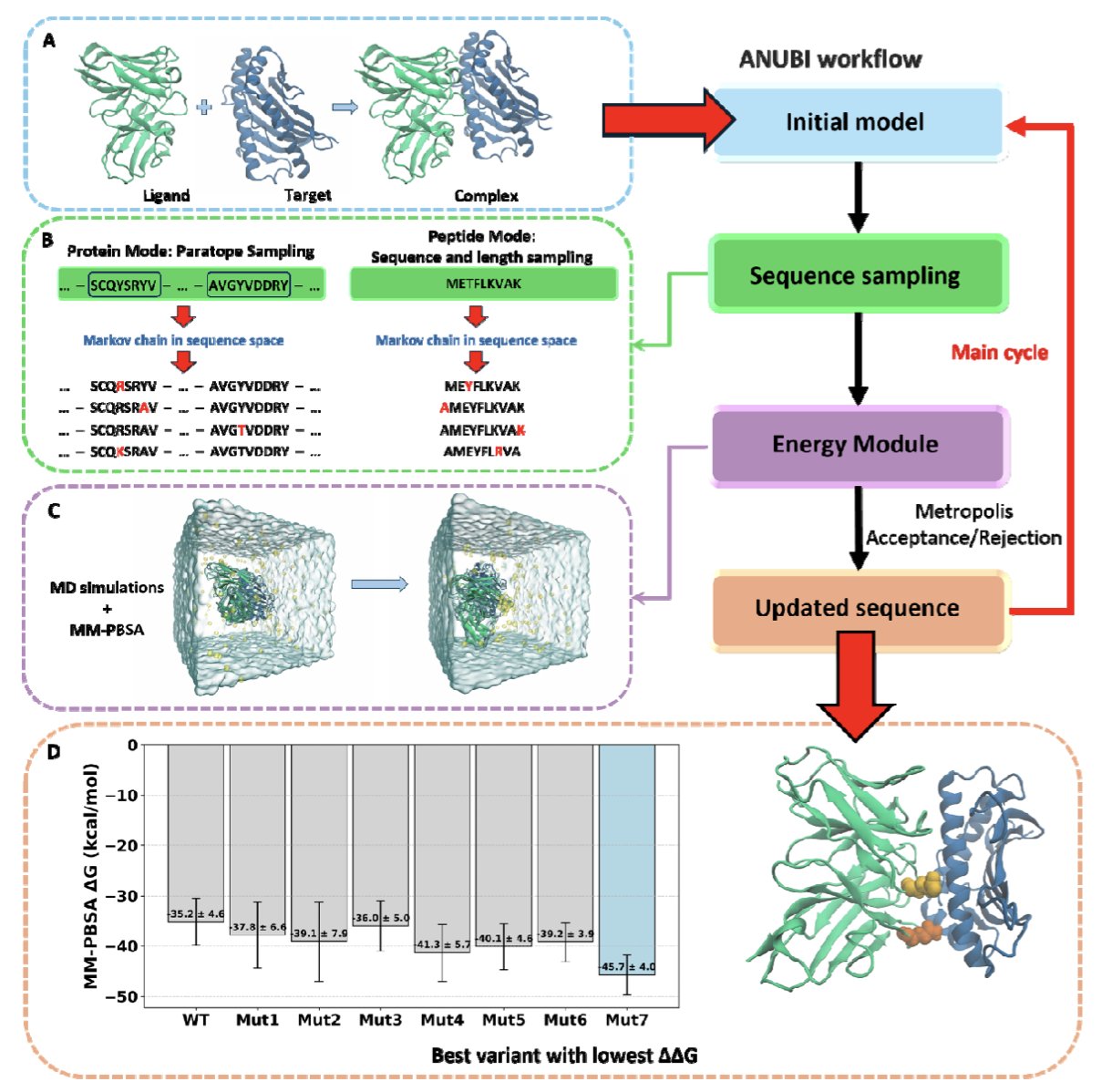
A key step in developing protein or peptide drugs is improving their binding affinity. This requires screening hundreds or thousands of amino acid mutations to find the “golden combination” that makes the drug bind more tightly to its target. This process is both time-consuming and expensive.
Researchers developed a software tool called ANUBI to simplify this workflow. You can think of it as a virtual molecular evolution engine.
Here’s how it works: first, the user provides the structure of a protein or peptide drug candidate. ANUBI then systematically attempts single amino acid substitutions—point mutations—in a specified region using a Monte Carlo method.
For each new mutation, the software evaluates its effect. This evaluation is the core of the process and the most computationally intensive part. ANUBI uses Molecular Dynamics (MD) simulations combined with the MMPBSA method to calculate the binding free energy.
MD simulations allow the molecule to move within the computer, searching for its most stable binding conformation. MMPBSA (Molecular Mechanics/Poisson-Boltzmann Surface Area) then uses snapshots from these dynamic simulations to estimate the binding affinity. MMPBSA is an established method; while its absolute calculations can have biases, it is effective at predicting trends and ranking candidate molecules.
If a mutation improves the binding energy, ANUBI keeps it and uses that new structure as the starting point for the next round of mutations. The entire workflow runs automatically once the user sets the initial parameters.
To validate its performance, the developers tested ANUBI in two scenarios: an antibody-antigen complex and a peptide-protein interaction. The results showed that after running for about 20 days on a single GPU, ANUBI identified mutants with a predicted binding energy improvement of about 20 kcal/mol.
It’s important to note that 20 kcal/mol is a very high theoretical value. In practice, achieving such a large increase in affinity with just a few point mutations is rare. MMPBSA calculations have their own error margins, so this number should be seen as a computational signal that these mutants have optimization potential. The actual effect must be verified by synthesizing the candidates and testing them with wet lab techniques like Surface Plasmon Resonance (SPR) or Isothermal Titration Calorimetry (ITC).
ANUBI’s value is that it packages a complex workflow, which would normally require a computational chemistry expert, into an easy-to-use tool. A drug discovery team can use it to screen dozens of promising candidates in a few weeks on a computer, at a low cost. They can then focus their lab resources on experimentally validating the most likely winners, saving time and money while making their experiments more targeted.
📜Title: Anubi: A Platform for Affinity Optimization of Proteins and Peptides in Drug Design 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.20.683353v1