
目录
- 屏蔽测试显示,MSA 列注意力机制是 OpenFold 的核心支柱,蛋白质长度决定了模型对几何推理与进化信息的依赖程度。
- 本研究整合多组学数据与机器学习,实现对基因网络及疾病标记物的高精度实时预测。
- KOSMOS 既能挖出 CDO1 这种真金,也会在 p53 这种基础题上翻车。严谨的「证伪审计」才是用好 AI 的关键。
- SculptDrug 利用贝叶斯流网络,通过感知蛋白表面边界和多层级结构特征,生成高亲和力且无空间位阻的类药配体。
- CNN 模型配合可解释性 AI(Explainable AI),实现 91.8% 的蛋白质功能分类精度。它自主「学会」关注组氨酸等关键催化残基,证明了算法的生物学合理性。
1. OpenFold 解剖:谁决定了预测精度?
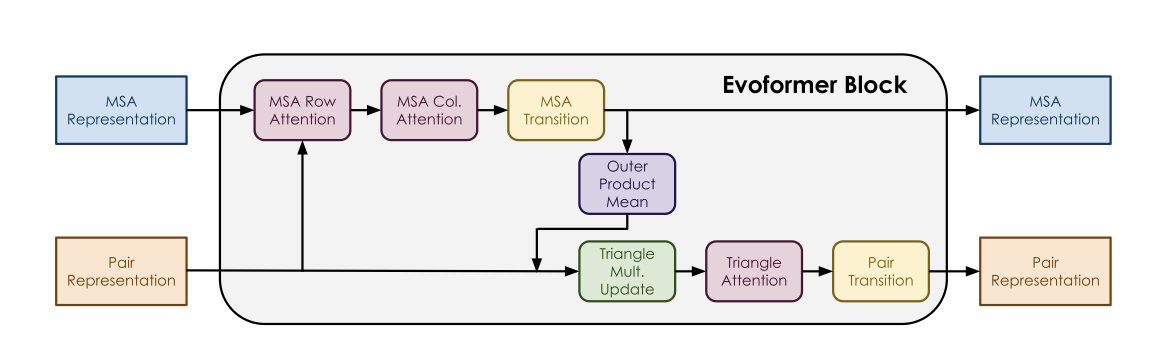
AlphaFold 和 OpenFold 虽已普及,但其内部运作对多数人而言仍是「黑盒」。佐治亚理工学院的 Tyler L. Hayes 和 Giri P. Krishnan 掀开了 OpenFold 的引擎盖,探究驱动这台机器的核心零件。
给模型做「基因敲除」
研究人员采用类似生物学「基因敲除」的直观方法:系统性跳过或「归零」Evoformer 模块中的特定组件,观察预测精度变化。实验使用 CAMEO 数据集中小于 700 个残基的蛋白质子集,确保结果覆盖各类常见蛋白结构。
MSA 是绝对主力
尽管模型复杂,MSA 列注意力机制(MSA Column Attention)、MLP 转换层及最终的配对(Pair)表示三个部分基本决定了预测准确度。
MSA 列注意力机制表现尤为突出。数据显示,对于许多蛋白质,仅保留该组件就能获得接近基准线的成绩。这印证了一个行业共识:现有的结构预测模型,本质是在提取多序列比对(MSA)中蕴含的进化信息。
长短蛋白「口味」各异
蛋白质长度影响其对模型组件的偏好:
优化方向
这项工作揭示了 OpenFold 的「思考」方式,指出了优化路径。已知某些蛋白主要依赖 MSA,通过优化该部分的计算效率,有望设计出轻量级模型。针对预测失败的短肽,应重点检查几何推理模块,而非盲目增加 MSA 深度。
📜Title: Quantifying the Role of OpenFold Components in Protein Structure Prediction
🌐Paper: https://arxiv.org/abs/2511.14781v1
2. 多组学+AI:高精度解析基因网络的新范式
- 多组学融合:整合基因组、转录组和蛋白质组数据,揭示隐藏的调控通路。
- AI 驱动高精度预测:利用机器学习显著提升基因功能和疾病标记物识别准确性。
- 动态实时监测:具备实时分析能力,捕捉细胞对药物或环境变化的动态响应。
药物研发常受困于复杂的基因网络。传统实验多聚焦单一靶点或通路,难以窥见全貌。这项研究试图构建全局视野。
作者开发的计算框架整合了基因组学(Genomics)、转录组学(Transcriptomics)和蛋白质组学(Proteomics)。这种全景视角将细胞运作的碎片拼接为连贯整体,揭示出以往隐匿的调控通路,直击细胞功能障碍的根源。
机器学习(Machine Learning)在此切实提升了预测精度。模型识别疾病标记物的可靠性优于传统方法,有助于降低早期药物筛选成本,提升诊断工具的精确度。
该框架具备实时分析能力。细胞对环境或药物的反应是动态的,此工具能实时监控基因网络变化。观察化合物介入后的网络扰动,能有效解析药物作用机理(Mechanism of Action)。
大量模拟和实验数据验证了该方法的鲁棒性。这一工具若普及,将为靶点发现与验证提供更清晰的指引。
📜Title: New Insights in Computational Biology: A Breakthrough Approach to Understanding Genetic Networks
🌐Paper: https://www.biorxiv.org/content/10.1101/2025.11.18.688681v1
3. KOSMOS 实测:AI 科学家是天才还是瞎蒙?
既然都在谈论「AI 科学家」能否独立搞科研,不如看看实战表现。这项研究将自主 AI 系统 KOSMOS 投入放射生物学(Radiation Biology)的深水区,要求它解决三个复杂问题。结果显示:它兼具惊人的直觉与初学者的笨拙。
挖掘金矿
KOSMOS 在预测细胞辐射反应时展现了敏锐的嗅觉。它提出假说:CDO1 基因是乳腺癌细胞辐射反应的关键预测因子。后续验证显示,数据确实支持这一观点。这正是我们对 AI 的期待:在海量数据中捕捉人类遗漏的线索,提供新的生物学视角。
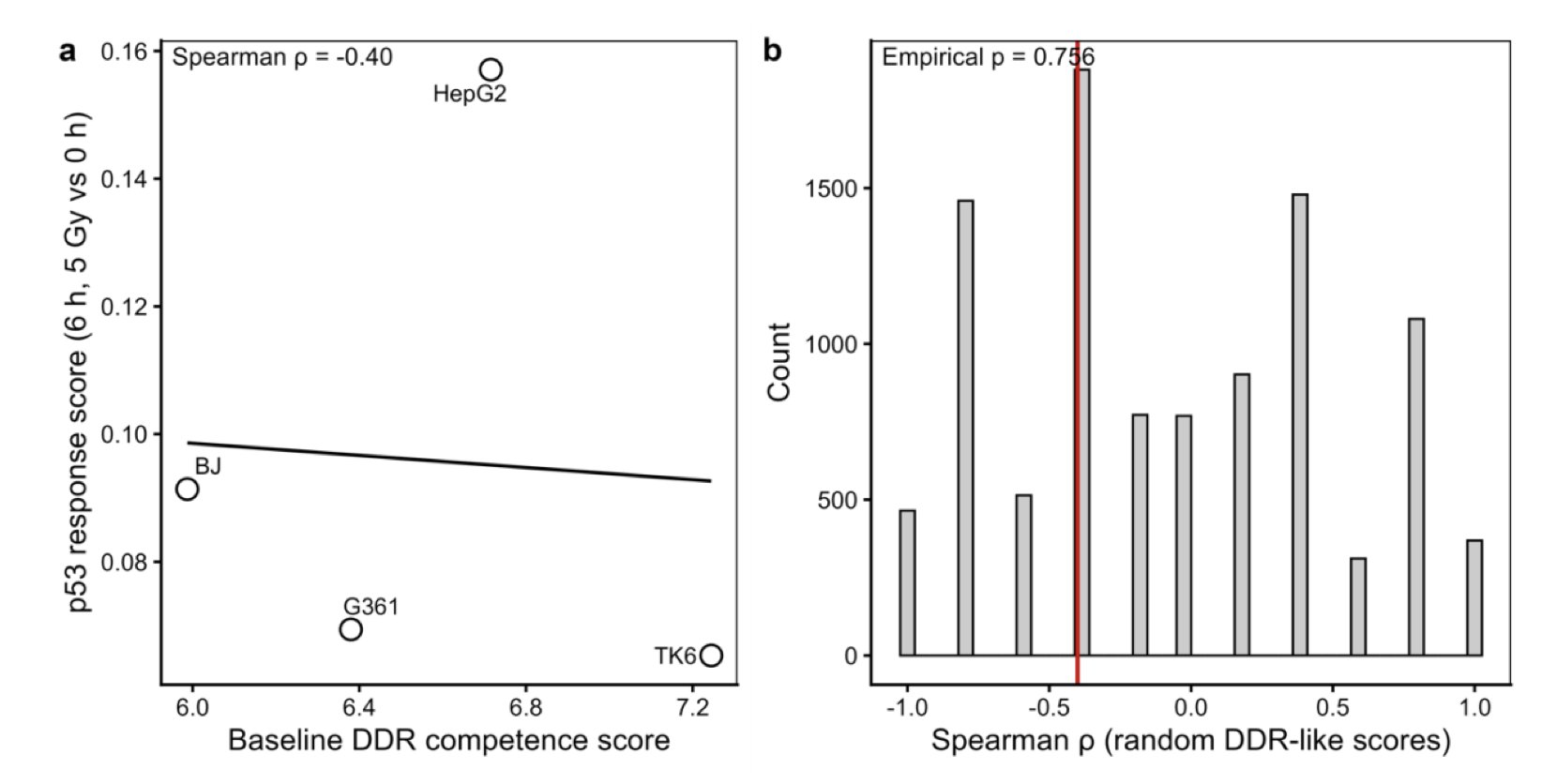
翻车现场
KOSMOS 试图将前列腺癌患者的治疗结果与基线 DNA 损伤反应及 p53 反应联系起来时,产生了「幻觉」。它信誓旦旦地生成了相关性假说,但真实数据表明这一关联根本站不住脚。
这给药物研发人员敲响警钟:AI 擅长发现模式,但不在乎模式是否具备生物学意义或统计学效力。盲信它的输出容易误入歧途。
证伪审计
论文的核心价值在于提出了一套「基于证伪的审计方法」(falsification-based auditing)。
核心逻辑是将 AI 生成的假说置于「零模型」(null models)中进行对抗测试。若假说无法在严格的统计学测试中击败随机模型或基础模型,就应果断舍弃。
行内视角
对药物发现从业者而言,KOSMOS 的表现并不意外。它像个精力旺盛却缺乏经验的初级研究员:高通量产出想法,天才洞见与胡扯混杂其中。
关键在于建立审计机制,而非单纯制造更「聪明」的 AI。必须用严谨的实验设计和统计学方法「审问」AI,才能将其转化为强有力的研发工具。
📜Title: When AI Does Science: Evaluating the Autonomous AI Scientist KOSMOS in Radiation Biology
🌐Paper: https://arxiv.org/abs/2511.13825v1
4. SculptDrug:用贝叶斯流「雕刻」完美配体
结构生物学家和药物化学家深知,基于结构的药物设计(SBDD)核心难点在于将分子「放进去」。许多生成模型产出的配体常与蛋白骨架发生严重立体位阻,或忽视口袋内的化学环境。
SculptDrug 正如其名,致力于在受限空间内「雕刻」出适配分子。
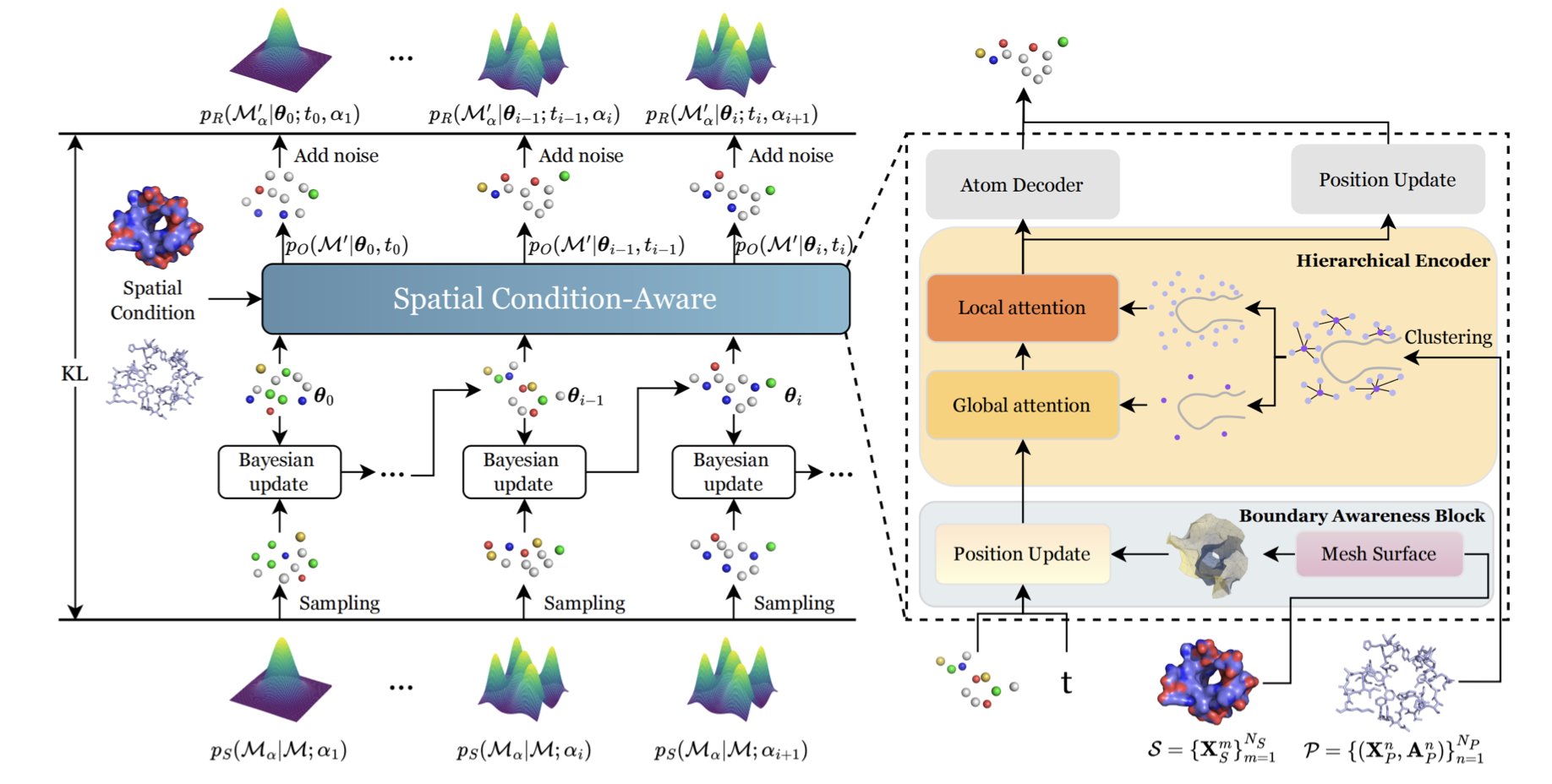
给生成过程装上「倒车雷达」
研究团队选用贝叶斯流网络(Bayesian Flow Networks, BFN)作为底层架构,以处理原子坐标等连续数据。核心创新在于边界感知模块(Boundary Awareness Block)。
若将蛋白表面视为墙壁,该模块将几何信息输入生成模型,强制其「看着路」生成配体。这有效降低了立体位阻(steric clashes),使分子顺应蛋白表面起伏,避免盲目填塞。
既看森林,也看树木
药物对接(Docking)中,结合口袋包含带电荷、疏水或极性的氨基酸残基,仅凭宏观轮廓无法对齐氢键或盐桥等关键相互作用。
SculptDrug 利用分层编码器(Hierarchical Encoder)破局。它一方面捕捉口袋全局形状以确保尺寸适配,另一方面深入微观,捕捉原子级环境。这种双尺度视野确保生成的配体形状匹配,化学性质也能与口袋内残基精确对接。
从噪声到药物的流变
生成策略采用渐进式去噪,类似精细打磨。模型从无序噪声起步,经迭代逐步调整原子位置和类型。这种细粒度控制保证了产物的空间几何构型合理,避免出现反直觉的长键长或扭曲键角。
实战表现
CrossDocked 数据集测试显示,SculptDrug 在结合亲和力(Binding Affinity)和类药性(Drug-likeness)上超越现有顶尖模型。消融实验证实,边界感知与分层编码器缺一不可。
目前 SculptDrug 仍将蛋白视为刚体。真实生物体内存在诱导契合(induced-fit),蛋白处于动态变化中,将动态构象纳入考量是未来的攻坚方向。但这一成果已向「AI 生成可用分子」迈出了坚实一步。
📜Title: SculptDrug: A Spatial Condition-Aware Bayesian Flow Model for Structure-based Drug Design
🌐Paper: https://arxiv.org/abs/2511.12489v1
5. XAI 让深度学习读懂蛋白质:告别「黑盒」
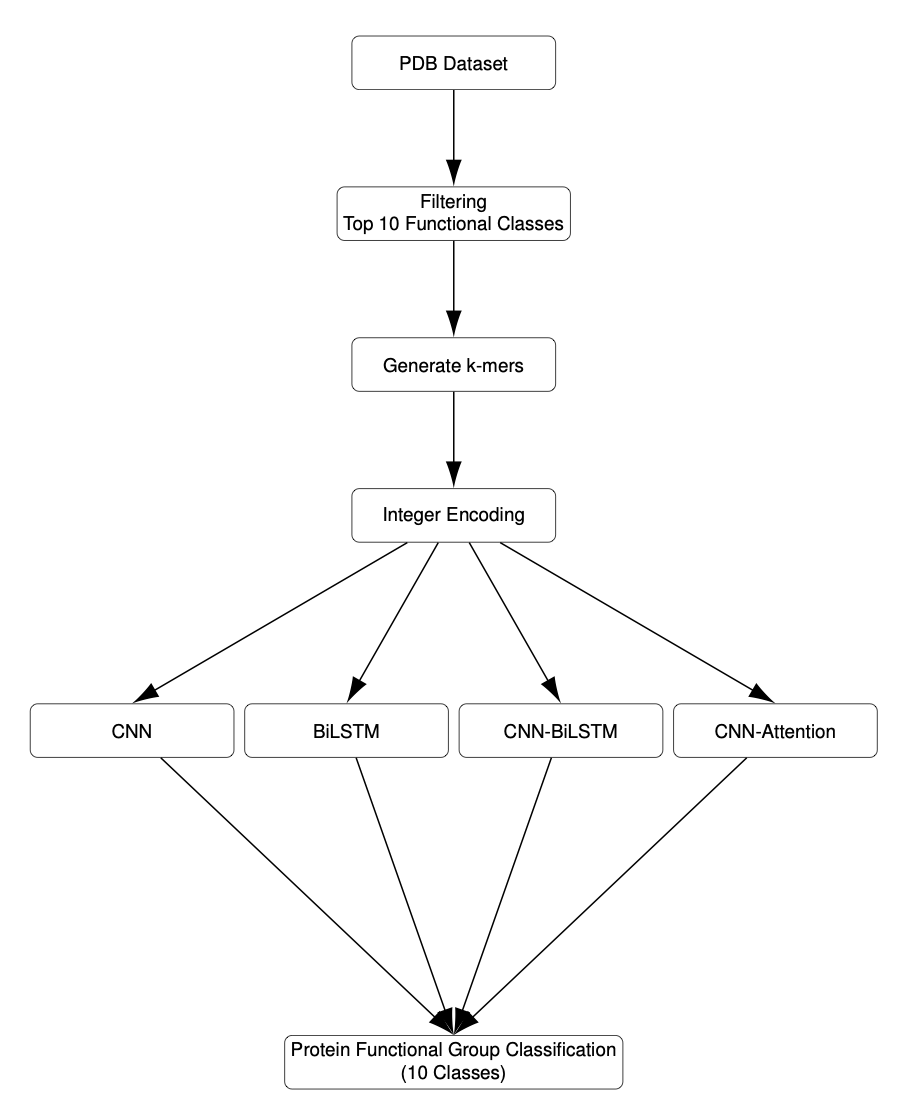
AI 辅助药物发现常面临信任危机:模型是真懂生物学,还是在玩弄统计数字?高准确率背后,判断依据往往成谜。这篇论文通过拆解「黑盒子」,揭示了深度学习模型的决策逻辑。
谁是赢家?
研究利用 PDB 数据库构建功能分类任务,对比 CNN、BiLSTM、CNN-BiLSTM 及带 Attention 的 CNN 四种架构。卷积神经网络(CNN)以 91.8% 的验证准确率夺魁。
蛋白质功能常由局部特定的氨基酸序列(基序)决定。CNN 的卷积核如同滑动的「显微镜」,擅长捕捉局部空间特征。处理长序列记忆的 BiLSTM 在此任务中关注点过于分散,难以锁定重点。
AI 看到了什么?
作者使用 Grad-CAM 和集成梯度(Integrated Gradients)两种技术,可视化模型的「注意力」。
模型锁定了组氨酸(His)、天冬氨酸(Asp)、谷氨酸(Glu)和赖氨酸(Lys)。
酶化学研究者对这些名字并不陌生。这些氨基酸是酶活性中心的「主力」,负责质子传递、金属离子结合及催化反应。AI 在无人为设定规则的情况下,自主发现了这些残基对蛋白质功能的重要性,其学到的特征与已知生化机制高度吻合。
架构的「性格」差异
XAI 揭示了不同模型的决策特征:
* Attention 模型激进,倾向将权重压在极短的子序列上,容错率低。
* BiLSTM 关注点遍布全序列,稀释了关键信号。
* CNN 表现平衡,能同时关注序列中多个局部基序,符合蛋白质多结构域协同工作的生物学现实。
在 AI 制药领域,仅追求高准确率不足以服众。XAI 确认模型关注真正的药效团或催化中心后,计算结果才能更放心地应用于湿实验。
📜Title: XAI-Driven Deep Learning for Protein Sequence Functional Group Classification
🌐Paper: https://arxiv.org/abs/2511.13791v1