
Table of Contents
- BioCG transforms biochemical reaction prediction into a constrained generation task, improving the model’s accuracy and efficiency in real-world scenarios.
- When data is sparse, using PCA to reduce the dimensionality of a VAE’s latent space before Bayesian optimization is an effective strategy for discovering new antimicrobial peptides.
- DeepChem now includes SE(3)-equivariant neural networks, simplifying the handling of 3D molecular geometry and making advanced modeling more accessible and reliable.
- SIGMADOCK uses a fragment-based SE(3) diffusion model, breaking ligands into rigid pieces for docking. This improves accuracy and efficiency, outperforming existing methods.
- LigifyDB is an interactive, pre-computed database that systematically predicts interactions between transcription factors and small molecules, providing a new tool for rapid biosensor development.
- The ID3 framework uses gradient optimization to design discrete mRNA sequences, solving key constraints more efficiently than traditional methods.
1. BioCG: Predicting Biochemical Reactions with Generative AI
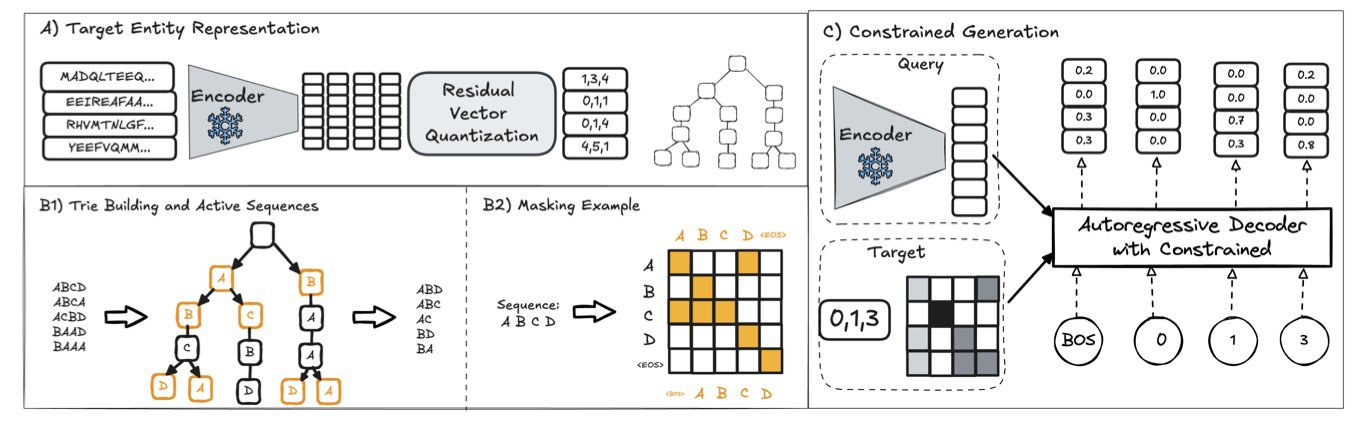
In drug development, predicting interactions between molecules—like drug-target interactions (DTI) or drug-drug interactions (DDI)—is a central challenge. Past AI models typically search for answers in a vast space of possibilities. This approach often leads to “hallucinated” results that don’t exist in the real world.
The BioCG model takes a new approach.
Researchers built an “answer library” of all known proteins and drugs. They redefined the prediction task as a generation task where the model must choose the correct answer from a finite set of options. This guarantees that any output from the model is a real biochemical entity.
To make this work, the researchers developed two key technologies.
The first is Iterative Residual Vector Quantization (I-RVQ). It assigns a unique identifier, made of a sequence of discrete numbers, to every known molecule (like a target protein). The model only needs to learn this identifier, not the molecule’s complex chemical structure, which simplifies the learning process.
The second is trie-guided constrained decoding. This technique guides the model in real time as it generates an identifier, ensuring every step moves toward a valid answer. If the model generates a sequence prefix that doesn’t exist in the answer library, the mechanism immediately corrects it. This focuses the model’s computational resources on distinguishing between meaningful biochemical options.
This new method performs well. In the classic BioSNAP DTI benchmark, when faced with proteins not seen during training, BioCG achieved an AUC of 89.31%, which is 14.3% higher than the previous best method. This shows that BioCG is more reliable in “cold-start” situations that resemble real-world research scenarios.
The researchers also designed an information-weighted training method that teaches the model to prioritize learning key decision points, allowing it to learn efficiently even with limited data.
BioCG’s shift in thinking improves prediction accuracy and makes the AI model’s generation process more reliable. Every step of its output can be verified, which is valuable in the rigorous field of drug discovery.
📜Title: BioCG: Constrained Generative Modeling for Biochemical Interaction Prediction 🌐Paper: https://openreview.net/pdf/e888694d2a8f7af2d08c2e095aab2d544eaae072.pdf
2. Compressing Latent Space with PCA: A New Way for AI to Design Antimicrobial Peptides
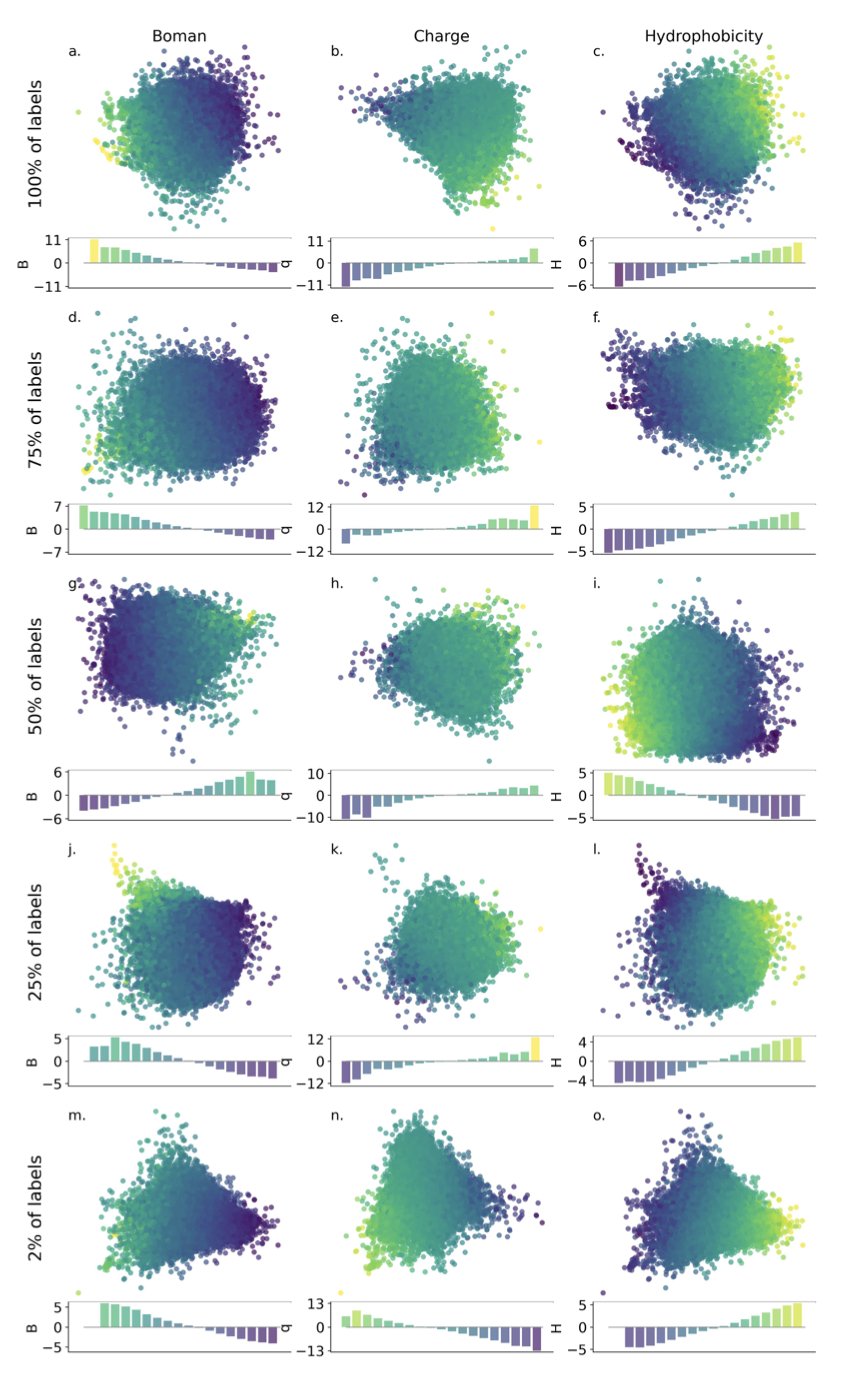
Drug discovery is a search through an enormous chemical space. For antimicrobial peptides (AMPs), the number of possible arrangements of just a few dozen amino acids is greater than the number of atoms in the universe. Finding an effective molecule here is like finding a needle in a haystack.
Recently, using generative AI models, particularly Variational Autoencoders (VAEs), to create a “map” of this chemical space has become a popular approach. A VAE can compress the vast space of peptide sequences into a low-dimensional, continuous “latent space.” You can think of this as a condensed map containing all the information about the peptide sequences. In theory, if you can find the “treasure” on this map and then decode it back into a specific peptide sequence, you have designed a molecule.
The next question is how to search this map efficiently. One common method is Bayesian Optimization. It acts like an explorer. Each time it tests a point (runs an experiment), it updates its understanding of the entire treasure map and then decides on the next most promising point to explore.
But the “map” generated by a VAE has too many dimensions and a complex topography. Running Bayesian optimization directly on it is like searching for the highest peak in a huge, fog-covered mountain range—it’s easy to get lost and inefficient.
Researchers used Principal Component Analysis (PCA) to simplify this complex map. PCA is like taking a complex 3D topographical map and flattening it into a 2D map while preserving key features like mountain ranges, peaks, and valleys. The researchers first used PCA on the high-dimensional latent space generated by the VAE, compressing it into a lower-dimensional, structurally simpler “projection space.” Then, they let the Bayesian optimization algorithm search for treasure on this simplified map.
This method has several advantages. First, the search space is smaller, which improves the optimization algorithm’s speed and its likelihood of finding the global optimum.
Second, this approach addresses a core problem in drug discovery: the scarcity of labeled data. Testing a molecule’s activity is time-consuming and expensive. With only a small amount of activity data, the latent space map learned by the VAE may be low-quality, causing different molecules to be jumbled together in a disorganized structure.
The study found that easily calculable physicochemical properties, like charge and hydrophobicity, can be used to pre-organize the latent space. This is like marking “water sources,” “forests,” and “deserts” on the map beforehand. Even without knowing the exact location of the treasure, these basic references make the search more directed. Experiments showed that organizing the latent space using “charge” led to the best performance for the subsequent Bayesian optimization.
This strategy remained effective even with only 2% of the data labeled.
When searching in the PCA projection space, the model also showed greater exploration. It didn’t just dig deeper in one promising area but was more willing to explore new, unvisited regions. In early-stage drug discovery, the ideal outcome is to find several structurally diverse families of promising candidate molecules, not a large number of similar molecules with mediocre effects. This broader exploration increases the chance of discovering entirely new molecular scaffolds.
This work shows that progress in AI for drug discovery doesn’t just depend on bigger, more complex models. Combining classic statistical tools like PCA to preprocess AI-generated representation spaces, and using basic chemical knowledge (like physicochemical properties), can solve problems efficiently with less experimental data. This approach is valuable for teams with limited resources or for projects exploring new drug targets.
📜Title: Semi-supervised latent Bayesian optimization for designing antimicrobial peptides 🌐Paper: https://arxiv.org/abs/2510.17569
3. DeepChem Integrates SE(3)-Equivariant Networks to Power 3D Molecular Modeling
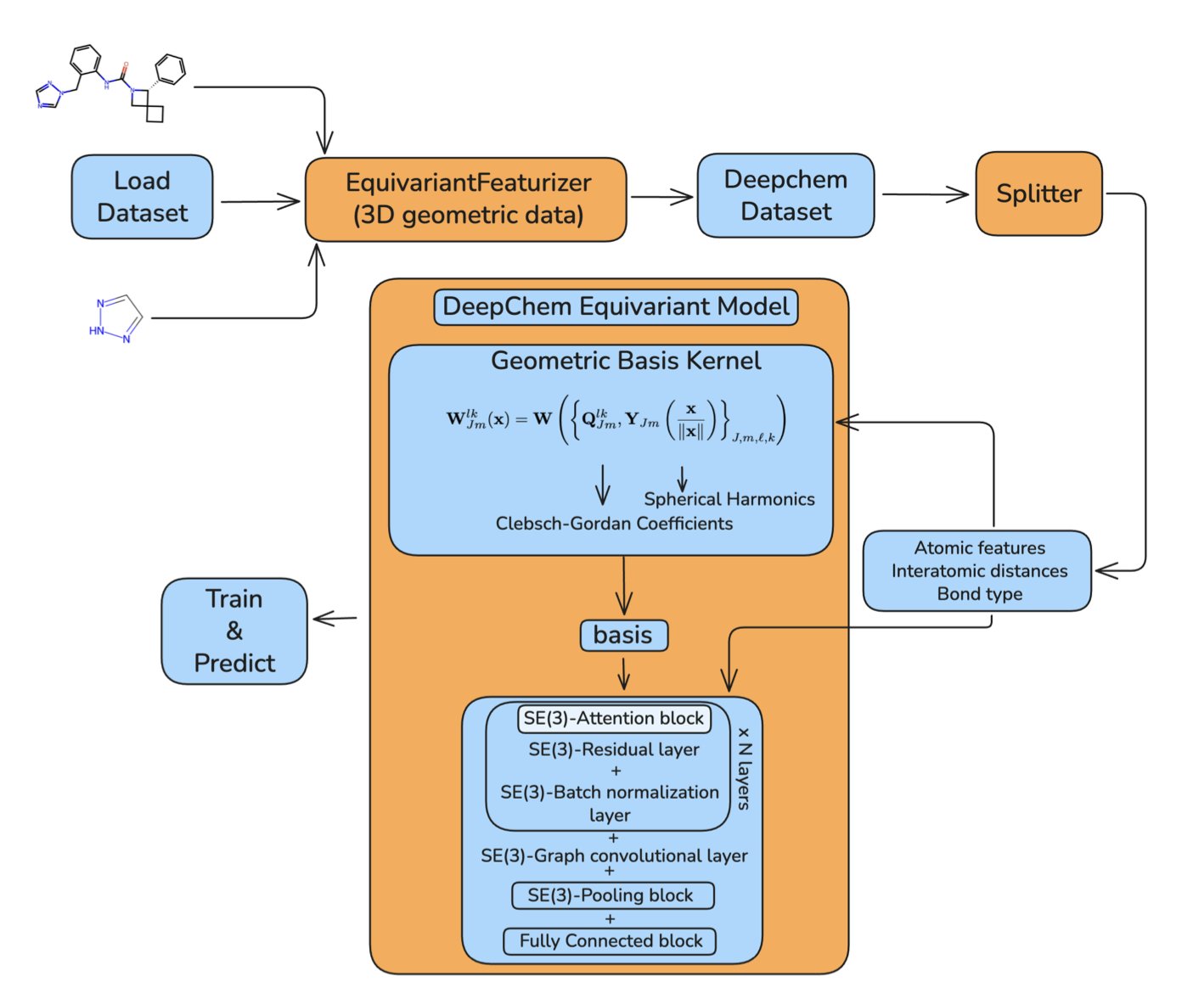
Handling the 3D spatial information of molecules is a major challenge in molecular simulation and drug discovery. A molecule’s physical properties, like energy or dipole moment, should not change when it is rotated or translated in space, but many machine learning models fail to understand this law. If you give a model the coordinates of a rotated molecule, it might mistake it for a new molecule and produce an incorrect prediction.
To address this, the open-source library DeepChem has integrated SE(3)-Equivariant Neural Networks.
SE(3) is the mathematical language that describes rotations and translations in 3D space. “Equivariant” means that when the input molecule is rotated, the features the network learns, such as the chemical environment of an atom, also rotate in a predictable way. Ultimately, the model can give consistent predictions for properties like energy that do not change with rotation. The model’s architecture itself follows the laws of physics.
The core technologies of the new module are spherical harmonics and irreducible representations. Spherical harmonics are like the building blocks for describing shapes on a sphere, similar to how a Fourier transform uses sine and cosine waves to construct a signal. The network uses these to efficiently encode and process the 3D spatial information around atoms and to ensure its behavior is correct under coordinate transformations. DeepChem has implemented classic models like the SE(3)-Transformer and Tensor Field Networks and has made them modular, so even non-experts in deep learning can use them.
The researchers tested the implementation on the standard QM9 dataset for predicting quantum chemical properties of small molecules. The results showed that the performance of the models in DeepChem is on par with top methods in the field, confirming the quality of the code. The models also incorporate attention mechanisms to better capture interactions between atoms.
Future optimization plans include introducing LieConv to improve computational efficiency and caching pre-computed basis functions to speed up training. These engineering considerations reflect a focus on computational efficiency and scalability for real-world applications.
📜Title: DeepChem Equivariant: SE(3)-Equivariant Support in an Open-Source Molecular Machine Learning Library 🌐Paper: https://arxiv.org/abs/2510.16897
4. SIGMADOCK: Rethinking Molecular Docking with a Fragment Diffusion Model
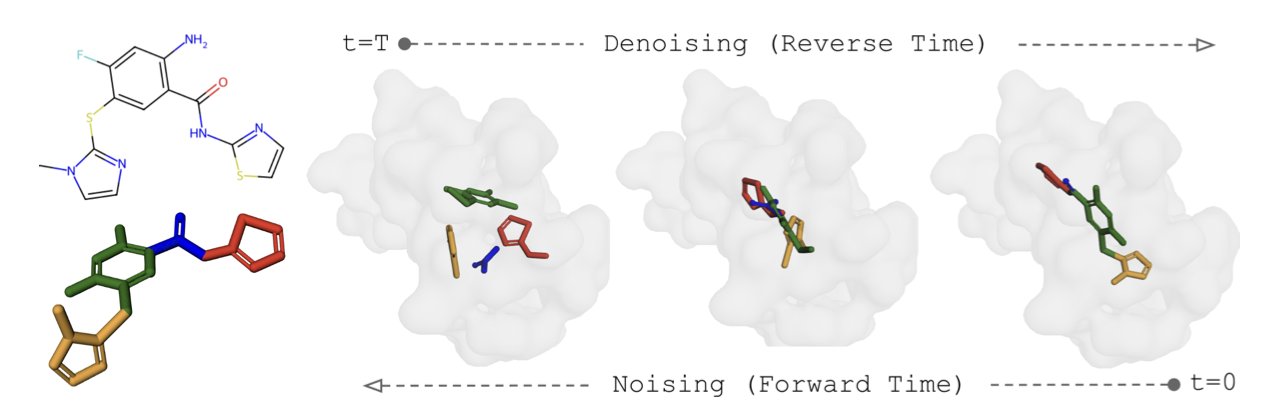
Molecular docking is a cornerstone of drug discovery. It finds drug molecules that can bind to a protein target. Traditional docking methods, whether based on physical force fields or deep learning models, all face the challenge of ligand flexibility. Ligand molecules have multiple rotatable chemical bonds, creating a vast and complex conformation space.
To solve this, some models try to generate the 3D coordinates of the entire molecule at once, but this is difficult and the results are often poor. Other models consider the molecule’s rotatable bonds, but the diffusion process for handling torsion angles is complex and the training dynamics are unstable.
SIGMADOCK offers a new idea: it breaks the ligand into multiple rigid fragments with fixed internal structures, which simplifies the problem. SIGMADOCK then applies an SE(3) Riemannian diffusion model to these fragments. The model first places the fragments randomly around the protein target and then gradually guides them to the correct position and orientation. This process uses geometric priors, making the learning process more efficient and stable.
The advantage of this method is that by breaking down the ligand, it reduces the number of degrees of freedom that need to be handled, which significantly lowers the computational complexity. On the PoseBusters benchmark, the model’s Top-1 success rate reached 79.9%, meaning that in most cases, the optimal conformation it predicted was closest to the true structure. This result not only surpasses other deep learning methods but also outperforms some classic physics-based docking software.
SIGMADOCK is highly efficient. It achieved high performance using less training data and computational resources, and it showed good generalization ability to unseen protein targets. This demonstrates the feasibility and efficiency of its method, setting a new benchmark for the reliability of deep learning in drug discovery.
📜Title: SIGMADOCK: Untwisting Molecular Docking with Fragment-Based SE(3) Diffusion 🌐Paper: https://arxiv.org/abs/2511.04854
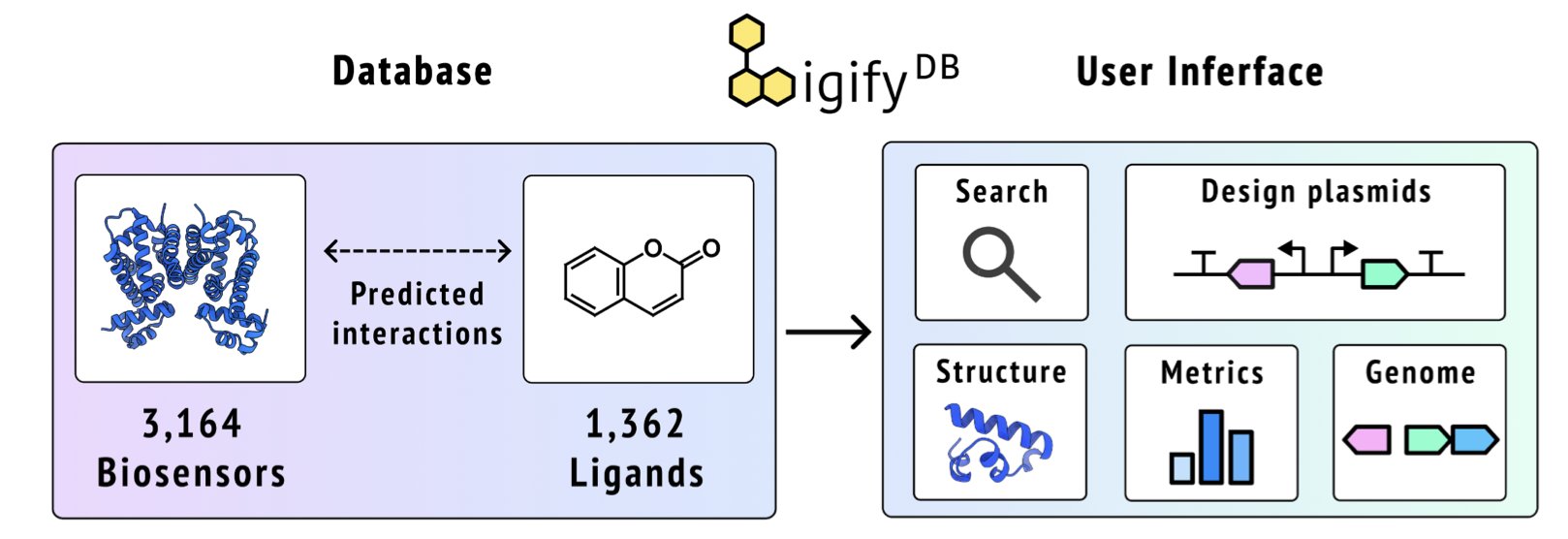
5. LigifyDB: A Fast Way to Discover Small-Molecule Biosensors
In synthetic biology and metabolic engineering, there is often a need for a “switch” that can sense a specific small molecule and regulate gene expression. This switch is called a biosensor. Finding a suitable biosensor is typically a time-consuming, trial-and-error process that requires searching through literature or screening from scratch.
A new tool called LigifyDB changes this. It acts like a massive, pre-computed “molecular matching directory.”
At the core of this directory are the relationships between small-molecule ligands and Transcription Factors (TFs). TFs are a class of proteins that can bind to specific sites on DNA and regulate gene expression. When a small-molecule ligand binds to a TF, it changes its conformation, which in turn affects its regulatory function. This is the working principle of a biosensor.
To build this directory, the research team started with the Rhea enzyme reaction database, a resource containing tens of thousands of known biochemical reactions. They systematically evaluated 10,965 small molecules from this database and used the Ligify workflow to predict the TFs that could potentially bind to them.
The key here is “pre-computation.” Traditional tools start calculating only after you submit a query, which means you have to wait. LigifyDB, on the other hand, has already calculated all possible combinations for 1,362 small molecules and 3,164 TFs. It’s like a dictionary with the definitions already written in the margins—you just have to turn to the right page to get the answer instantly.
The LigifyDB website is more than just a data list. When you search for a small molecule (like an amino acid), the system returns a list of TFs that might bind to it. Clicking on one of them shows detailed information.
The platform also has a built-in plasmid designer.
After finding a potential sensor, it must be validated in the lab. This designer can quickly generate the gene sequence for the reporter plasmid needed for the experiment. Users can choose a promoter and a reporter gene (like Green Fluorescent Protein, GFP), and the tool automatically inserts strong terminators and other regulatory elements to ensure the sensor circuit is stable and modular. Once the design is complete, you can directly download a GenBank file for gene synthesis. This feature connects computational prediction with experimental validation.
The database can be continuously expanded. As enzyme databases are updated and prediction algorithms improve, LigifyDB’s content will become richer and more accurate. For researchers who need to find a sensor for a specific metabolite, it is a valuable resource.
📜Title: LigifyDB: An interactive platform for predicted small molecule biosensors 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.20.683484v1
6. A New Paradigm for mRNA Optimization: Using Gradient Descent for Sequence Design
Designing an mRNA sequence that is stable and expresses efficiently is like searching for the optimal solution in an astronomical codebook made of four letters: A, U, C, and G. The goal is to find a sequence that not only translates into the target protein efficiently but also remains stable. Traditional methods like genetic algorithms are like searching for treasure blindfolded—they are inefficient and cannot guarantee finding the best solution.
The ID3 framework brings a new approach to this problem.
How can gradient descent handle a discrete sequence of bases?
Gradient-based methods are powerful optimization tools. They are like a blind person walking down a hill, always choosing the steepest path to reach the bottom efficiently. But this method requires a continuous, smooth “slope.” An mRNA sequence, made of discrete bases, is like a series of “steps,” so a gradient cannot be calculated directly.
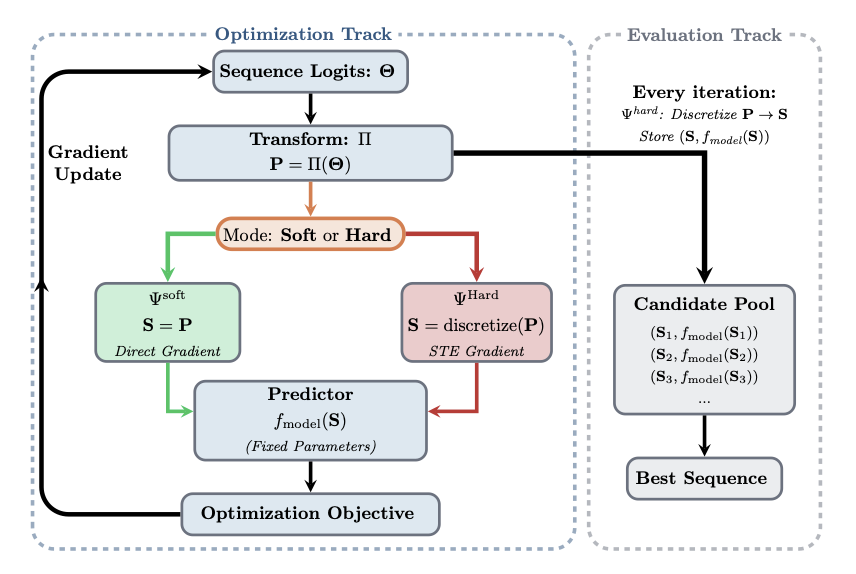
The core idea of the ID3 framework is to build a smooth slope for this blind person.
It does this through a “decoupled optimization-evaluation” architecture. The optimizer directly adjusts the “probability distribution” of codons at each position, rather than manipulating the discrete A/U/C/G sequence. For example, for a position that encodes leucine, the optimizer adjusts the respective probabilities of the six synonymous codons. These continuous probability values form a smooth optimization space where gradient descent can be applied.
Next, the evaluation module samples specific, discrete mRNA candidate sequences based on this probability distribution. These sequences are fed into existing deep learning models to predict their stability or expression efficiency. The prediction results are then fed back to the optimizer to guide the next adjustment of probabilities.
How does it ensure the protein sequence doesn’t go wrong?
While optimizing the mRNA, one strict rule must be followed: the final translated amino acid sequence must be correct. If the encoded amino acid is changed just to improve mRNA stability, the entire molecule becomes useless.
To prevent this, the authors designed three constraint mechanisms:
- Codon Profile Constraint: This is a preset preference that guides the optimizer to favor certain specific codons.
- Amino Matching Softmax: This is the key line of defense. It forces probabilities to be assigned only to synonymous codons that encode the same amino acid. The probabilities for all other codons are set to zero. This ensures the optimizer only chooses from allowed options and does not select the wrong amino acid.
- Lagrangian Multipliers: This mathematical tool handles hard constraints by introducing a penalty term. Any optimization attempt that tries to change the amino acid sequence is penalized and thus corrected.
Why is it better?
In tasks optimizing mRNA accessibility and the Codon Adaptation Index (CAI), ID3 outperformed traditional methods like genetic algorithms and simulated annealing. It found the global optimum faster and more accurately.
The ID3 framework can be directly connected to existing deep learning prediction models without retraining, which lowers the barrier to entry and speeds up practical application.
The ID3 framework also comes with a rigorous proof of convergence. In computational biology, many algorithms are effective but lack theoretical backing. The mathematical guarantee of ID3 makes its results more reliable.
The ID3 framework provides an efficient and reliable computational tool for mRNA sequence design. It solves the conflict between discrete sequences and continuous optimization and has the potential to accelerate the discovery of high-performing molecules in the development of mRNA vaccines and therapies.
📜Title: Gradient-based Optimization for mRNA Sequence Design 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.22.683691v1