
Table of Contents
- DMol uses a graph diffusion model and motif compression to generate molecules 10 times faster, preserving key chemical motifs and excelling in both efficiency and chemical validity.
- The H3-DDG model accurately predicts changes in protein binding free energy (∆∆G) after mutation by simulating complex multi-level, many-body interactions, performing especially well for multi-point mutations.
- A Bayesian learning framework offers a new approach to optimizing molecular force fields, using probabilistic methods to improve accuracy and robustness, particularly for charged, complex biological systems.
- The MolTD model boosts 3D molecule generation speed by nearly 100 times by optimizing atom arrangement and feature adjustment, while ensuring high-quality molecular structures.
- AlphaFold 3’s structure prediction scores can effectively distinguish between good and bad gene models, providing a new tool for annotating genomes that lack experimental data.
1. DMol: Generate Molecules 10x Faster While Keeping Key Chemical Motifs
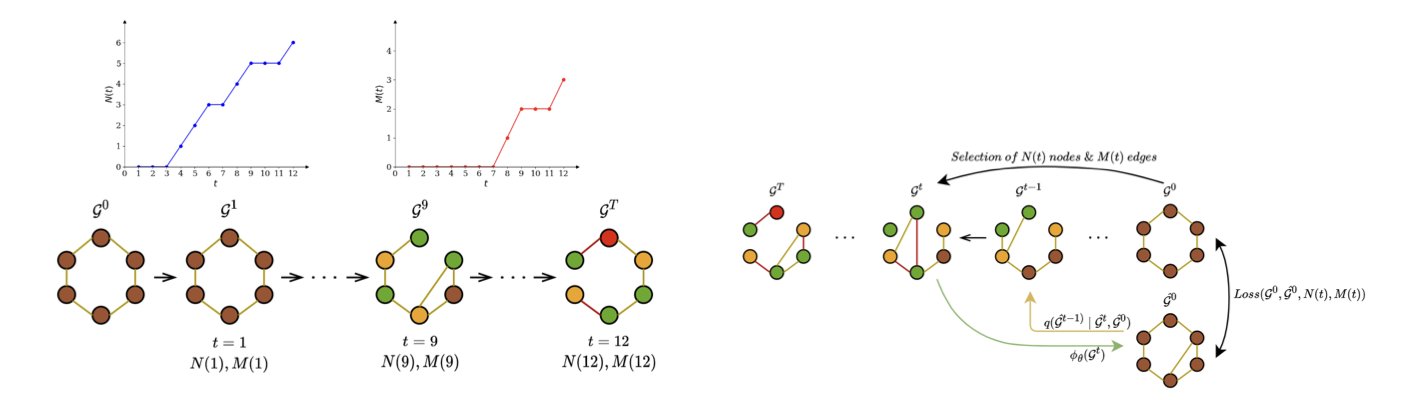
The field of drug discovery is always looking for faster and better ways to design new molecules, and generative models offer a new path. Graph diffusion models are a popular choice right now, but they are generally slow. Generating a single molecule from scratch can take hundreds or even thousands of diffusion steps. For drug development, which requires screening huge numbers of molecules, this is a bottleneck.
The DMol platform was built to solve this problem. Its core idea is to drastically reduce the number of diffusion steps without sacrificing molecular quality. DMol cuts the process down to just a few dozen steps, making it about 10 times faster than mainstream methods.
Two key innovations make this possible.
First, DMol uses a hybrid approach to adding noise. Traditional diffusion models typically learn to restore a molecule’s structure after randomly corrupting it. DMol combines both deterministic and random corruption methods. This gives the model better control over the diffusion process, allowing it to learn how to generate valid molecules in fewer steps.
Second is a technique called “motif compression.” Molecules are made of many chemical motifs that have specific functions, like benzene rings or carboxyl groups. These motifs define a molecule’s basic properties.
DMol identifies these chemical motifs and “compresses” them into a single “supernode.” In the diffusion model, a complex benzene ring is treated no differently than a simple carbon atom. This way, these important functional groups remain intact while the model generates the molecular backbone. Finally, the model “unpacks” these supernodes back into their original motif structures.
This approach simplifies the model’s learning task, improves computational efficiency, and ensures the chemical validity of the generated molecules. Experiments on standard industry datasets like QM9 and MOSES confirmed this. Molecules generated by DMol show excellent performance in validity, uniqueness, and novelty. Metrics like quantitative estimate of drug-likeness (QED) were 4% higher than existing models like DeFoG and DiGress.
DMol also supports conditional generation based on a molecular scaffold. A researcher can provide a core structure, and the model can “grow” new molecules with specific properties from it. This is very useful in drug optimization.
DMol increases the speed of molecule generation and, through motif compression, ensures chemical accuracy. A tool that balances speed and quality is very attractive to researchers on the front lines of development.
📜Title: DMol: A Highly Efficient and Chemical Motif-Preserving Molecule Generation Platform 🌐Paper: https://openreview.net/pdf/0f1d8aa866eb76c5f8a65f0847079723262ef2aa.pdf
2. H3-DDG Model Accurately Predicts Protein Mutations to Aid Drug Design
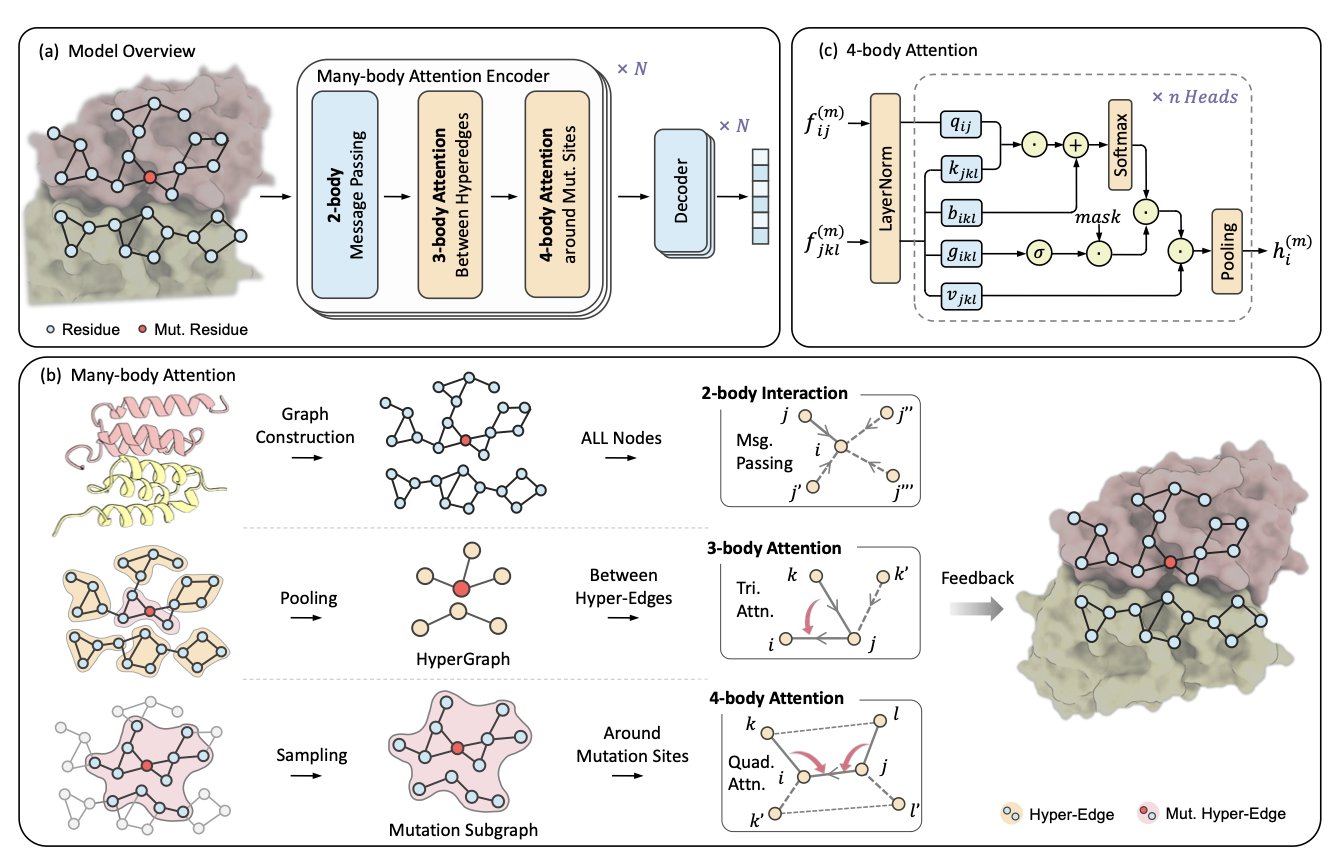
Predicting the change in binding free energy (∆∆G) caused by protein mutations is a central challenge in protein engineering and drug development. Swapping just one amino acid can alter a protein’s binding affinity, determining whether a drug molecule succeeds or fails. Traditional computational methods struggle to handle the complex network of interactions inside a protein, especially when multiple amino acids mutate at once.
Researchers have developed a new computational framework called H3-DDG to tackle this challenge. The method treats a protein as a complex, multi-level system.
First, H3-DDG builds a hypergraph to represent the protein’s structure. A hypergraph can describe higher-order interactions, such as a hydrogen bond network formed by a water molecule connecting two amino acid residues, or π-π stacking formed by several aromatic rings. This is like upgrading from a 2D social network map to a 3D model that can show interactions within small groups of people, providing richer information.
Next, the model uses a hierarchical many-body attention mechanism to examine the effects of a mutation at different scales. It can focus on changes in the chemical environment within a few angstroms (Å) of the mutation site, and it can also observe how this local change propagates through a chain of interactions to affect another functional region dozens of angstroms away. Accurately predicting multi-point mutations requires considering both fine-grained local structures and global long-range effects, as the impacts of multiple mutations are not simply additive.
H3-DDG’s predictive power was validated on several industry-standard datasets. On the SKEMPI v2 dataset, its predictions had a Pearson correlation of 0.75 with experimental data. For multi-point mutations, it improved upon the previous best method by 12.10%. On the more challenging BindingGYM dataset, its performance improved by 34.26%.
To demonstrate the validity of the model’s design, the researchers conducted a series of ablation studies. The results showed that removing the many-body attention mechanism caused a 5.4% drop in prediction accuracy, highlighting that accurately capturing higher-order interactions like hydrogen bond networks is crucial to the model’s success.
Theoretical success needs to be applied in practice. The researchers used H3-DDG to optimize an antibody targeting the SARS-CoV-2 virus. The model’s predictions helped identify mutations that could enhance the antibody’s binding to the viral spike protein, providing guidance for designing more effective therapeutic antibodies. This shows H3-DDG’s potential to accelerate drug discovery and protein engineering.
The researchers also noted that H3-DDG still needs to be tested on larger datasets and integrated with high-throughput experimental workflows to be useful in diverse, real-world R&D scenarios.
📜Title: Accurately Predicting Protein Mutational Effects via a Hierarchical Many-Body Attention Network 🌐Paper: https://openreview.net/pdf/d224adc934a88117d29e0bbc68a2334d4b96fdbb.pdf
3. Bayesian Learning: The Future of Molecular Force Fields?
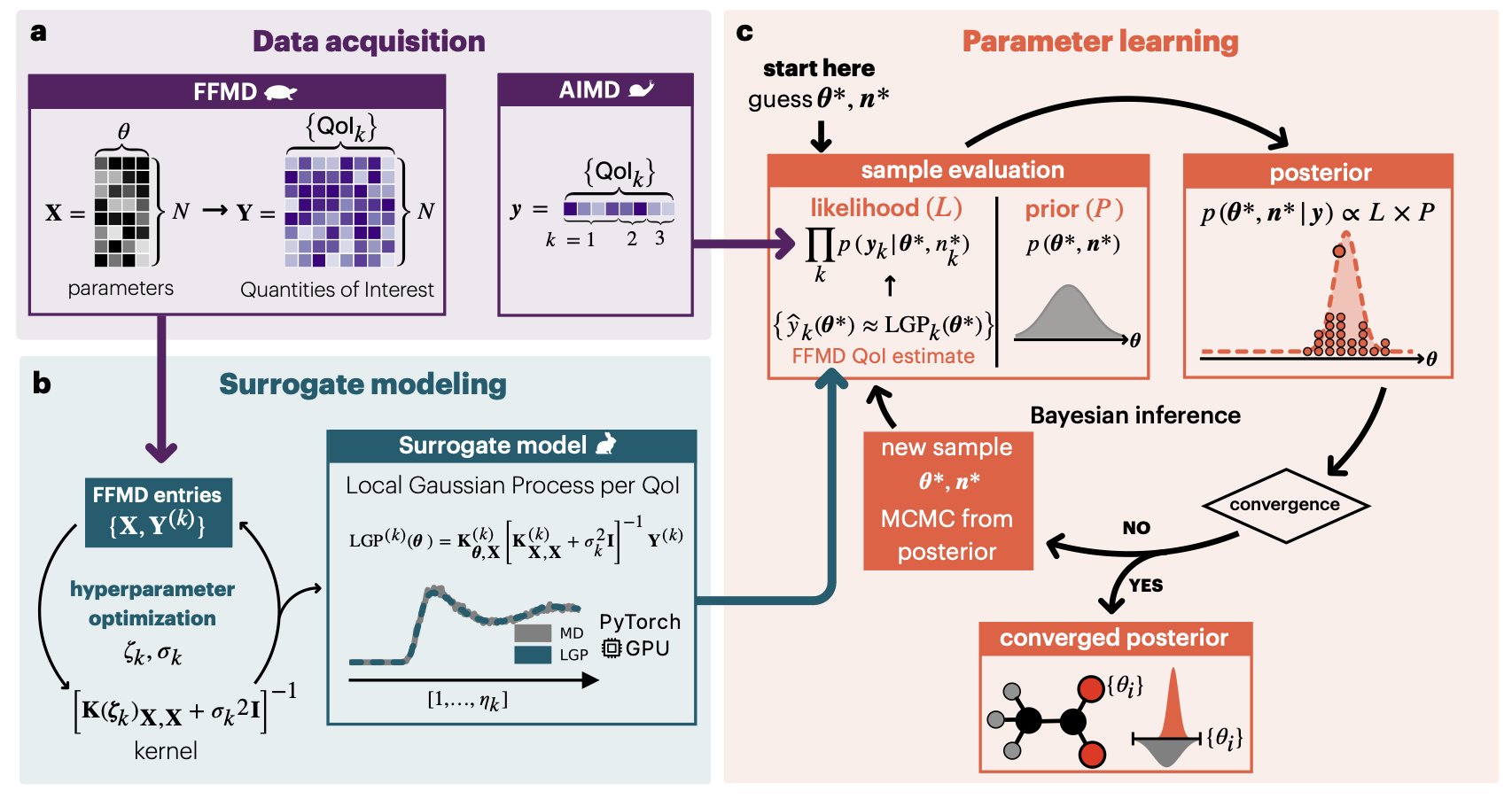
Force fields are the foundation of molecular simulation. Traditional force field development relies on experience and trial-and-error, making the process opaque and the results often suboptimal, especially when dealing with complex biological macromolecules. A new study proposes a Bayesian learning framework to make force field development more scientific and transparent.
The method treats both force field parameters and experimental data as probability distributions, which allows it to quantify the model’s uncertainty. The result is a set of possible parameter combinations and their corresponding probabilities. This is like watching a film that shows all possible states of a system, rather than looking at a single static photo.
The researchers demonstrated the framework’s capability using 18 biologically significant molecular fragments, including components of proteins, nucleic acids, and lipids. They found that the optimized parameters performed better than existing force fields when describing charged systems.
To prove the method’s practicality, they simulated the binding of calcium ions to cardiac troponin C, a key step in heart muscle contraction. Traditional simulations struggle to accurately capture this interaction. The Bayesian method provided more accurate simulation results and, by using multiple parameter sets, revealed the robustness and variability of the model’s predictions.
To address the high computational cost associated with probabilistic methods, the researchers used local Gaussian processes from machine learning to speed up the Bayesian inference. This makes the method computationally feasible for complex macromolecular systems.
Transferability is key in drug development. The study confirmed that parameters optimized on small molecular fragments could be successfully transferred to larger biomolecular systems. They verified this by simulating aqueous solutions and protein-ligand interactions, demonstrating the framework’s potential for broader applications.
📜Paper: Bayesian Learning for Accurate and Robust Biomolecular Force Fields 🌐Paper: https://arxiv.org/abs/2511.05398v1
4. MolTD Model Speeds Up 3D Molecule Generation by Nearly 100x
Drug discovery requires getting high-quality 3D molecular structures quickly. For current generative models, speed is a bottleneck when handling the 3D geometry of molecules. A new method called MolTD, developed by a team at Tsinghua University, accelerates this process.
The speed bottleneck in 3D molecule generation lies in determining the arrangement of atoms. Only after that can their coordinates and chemical features be refined.
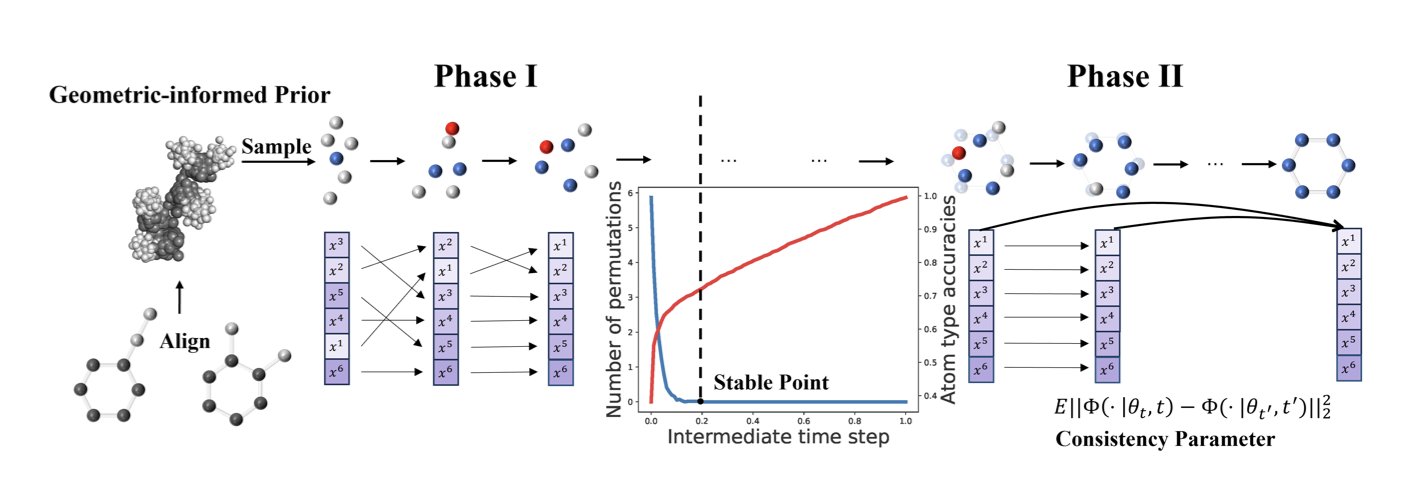
MolTD speeds up the process by splitting generation into two steps.
The first step is Permutation. Instead of predicting from scratch, the model references representative molecular structures from a dataset. This “geometric-informed prior” helps the model plan the general positions of atoms more quickly, forming a stable molecular backbone and reducing the number of computational steps needed to reach a stable structure.
The second step is Adjustment. Once the molecular backbone is in place, the details of each atom need to be refined. For this, the researchers designed a “consistency parameter objective,” a technique that makes the process of adjusting atomic features faster and more accurate.
On the QM9 and GEOM-DRUG datasets, MolTD achieved top performance in just 10 sampling steps. This is about 7.5 times faster than previous models and nearly 100 times faster than mainstream diffusion models. This speed increase is significant for drug development, which often requires generating and screening molecules on a large scale.
MolTD is widely applicable. It can achieve better results with less computation in both de novo drug design and target-based drug design tasks. This shows its potential for real-world applications like drug discovery and new materials design.
📜Title: Accelerating 3D Molecule Generative Models with Trajectory Diagnosis 🌐Paper: https://openreview.net/pdf/9b6d2adfb4ba64950f7ce5bdfa7c078e3013c8a7.pdf 💻Code: https://github.com/GenSI-THUAIR/MolTD
5. Are AlphaFold Scores a New “Gold Standard” for Gene Models?
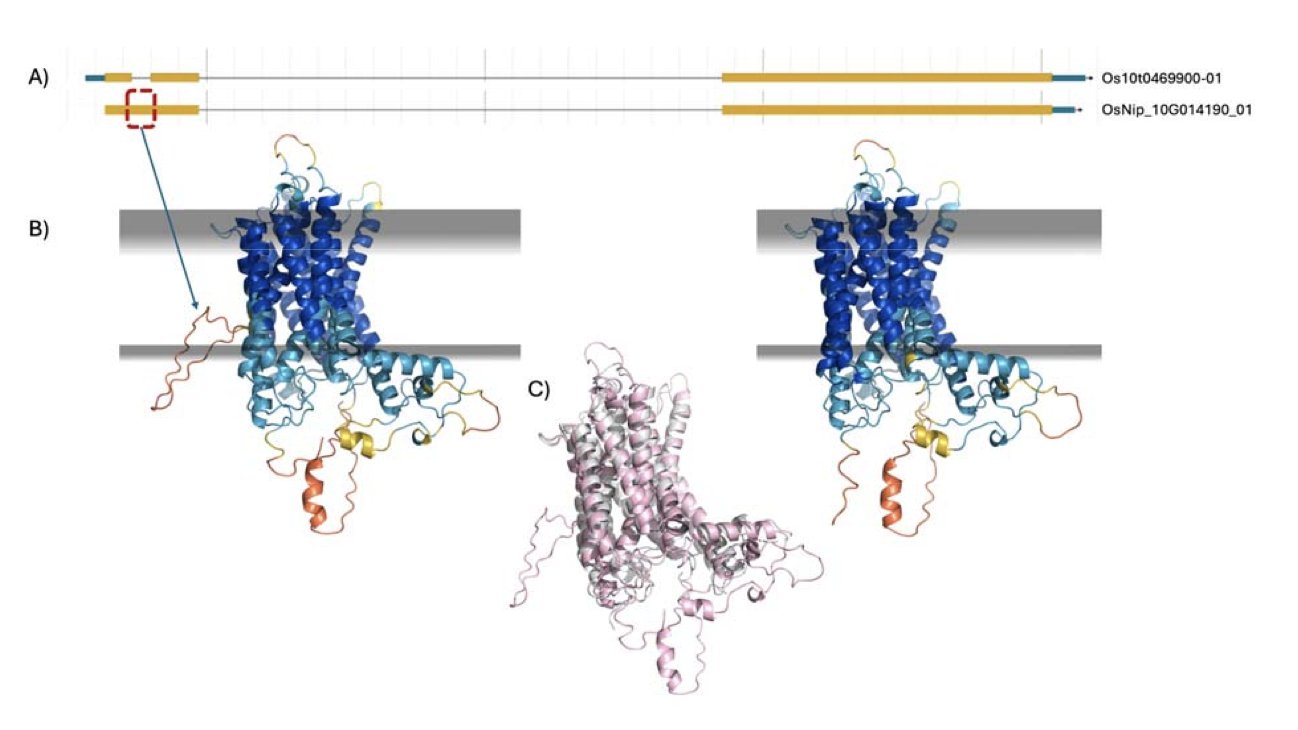
Genome annotation means marking the locations and boundaries of genes on a long DNA sequence. Automated processes are fast but often make mistakes. Manual curation is accurate but extremely slow, making it impractical for the vast amount of sequencing data from new species.
A new approach uses a basic principle of biology: a correct gene model, when translated into an amino acid sequence, should fold into a stable 3D protein structure. An incorrect model will produce a polypeptide chain that can’t fold properly. AlphaFold is a tool that can determine how well a protein structure folds.
Using Structure Prediction as a Referee
Researchers tested this idea on three species of fungi and protozoa, for which a large amount of expert-curated gene data exists. They compared automatically generated gene models with the expert-refined ones, having AlphaFold 3 predict the corresponding protein structures for each and assign a score.
The results showed that AlphaFold 3’s scores consistently favored the expert-refined models.
This is equivalent to using a physicochemical principle—whether a protein can fold correctly—as a standard for judging the quality of a gene annotation. And its judgment aligns closely with that of human experts.
A Combination Approach Works Better
Combining AlphaFold 3 with the structure alignment tool Foldseek can further improve the accuracy of screening gene models.
Here’s how it works: 1. AlphaFold 3 provides a score: It assesses whether an amino acid sequence can fold into a reasonable structure. 2. Foldseek performs a comparison: It compares the predicted structure against a database of known protein structures to find similarities.
If a protein translated from a gene model gets a high score from AlphaFold (meaning its structure is plausible) and Foldseek can find a similar homolog in its database, the model is very likely to be correct. This combined strategy is like authenticating an artifact: you check its craftsmanship and also verify its style and origin.
A Lingering Challenge: Disordered Regions
The main challenge for this method comes from Intrinsically Disordered Regions (IDRs) in proteins.
These regions do not have a fixed 3D structure under physiological conditions. AlphaFold is designed to find stable structures, so it gives these regions low scores. This could cause a correct gene model to be wrongly flagged as low-quality simply because its protein contains long IDRs.
The next key problem to solve is how to teach the model to distinguish between an “incorrect sequence that can’t fold” and a “correct sequence that is naturally disordered.”
For the massive number of newly sequenced genomes that lack manual curation, this automated evaluation method based on protein structure has the potential to improve the overall quality of gene data. High-quality gene data is the foundation for all subsequent functional research, including the discovery of drug targets.
📜Title: The Promise of AlphaFold for Gene Structure Annotation 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.21.683479v1