
Table of Contents
- Researchers from Fudan University developed Pro4S, a multi-modal model that fuses protein sequence, structure, and surface features to predict protein solubility more accurately, providing an efficient tool for designing and screening highly expressed proteins.
- In molecular property prediction tasks, Large Language Models (LLMs) handle InChI and IUPAC names better than traditional SMILES, possibly because InChI and IUPAC contain more explicit atomic information and appear more frequently in training data.
- The SPECTRA framework “interpolates” molecules in the spectral domain, creating chemically valid but non-existent molecules to fill sparse data regions and improve prediction accuracy in key property ranges.
- The POEMS model uses sparse decoding and a Product of Experts design to solve the interpretability challenge in multi-omics data integration while maintaining predictive performance, making it possible to discover cross-omics biomarkers.
- RBPtool combines a large language model with geometric deep learning to accurately predict RNA-protein binding and can also design RNA molecules in reverse for specific targets.
1. Pro4S: Fusing Sequence, Structure, and Surface Information for AI-Powered Protein Solubility Prediction
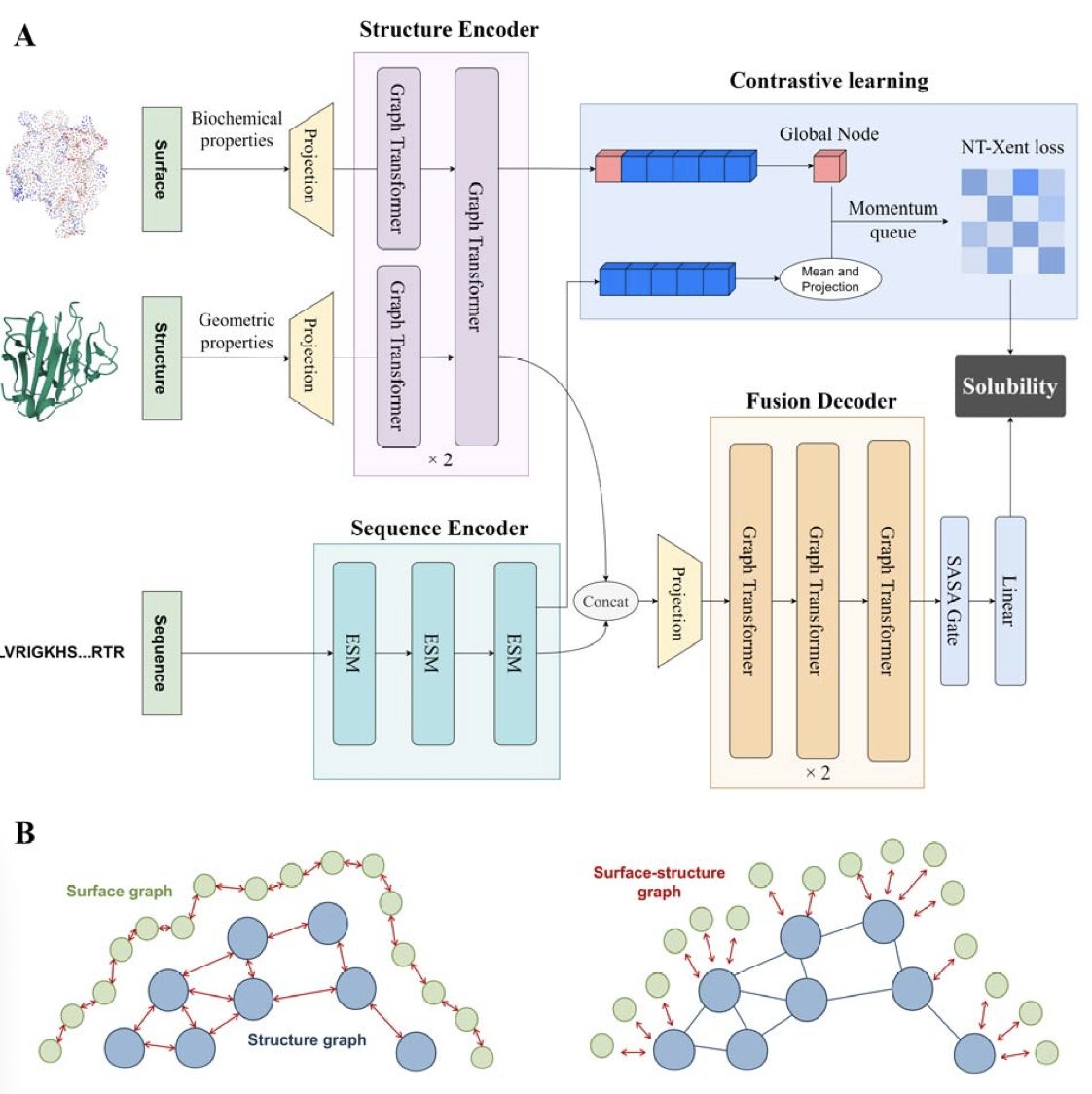
Protein solubility is a critical factor in protein-based projects. If a protein precipitates after expression, it’s difficult to move forward with functional studies or structural analysis. The traditional trial-and-error approach to screen for highly soluble mutants or sequences is both time-consuming and labor-intensive. Computational methods offer a new path, but their predictive accuracy needs improvement. The Pro4S model, recently developed by a team at Fudan University, offers a new solution.
The model’s core idea is to synthesize information from multiple dimensions. Many existing methods focus only on the amino acid sequence or the 3D structure, failing to use all available information. Pro4S performs a comprehensive analysis of the protein by processing three levels of data at once:
- Sequence Information: It uses the ESM Large Language Model to extract evolutionary and functional patterns from the protein sequence.
- Structural Features: It uses Graph Neural Networks to analyze the protein’s 3D structure, such as the distances and angles between atoms.
- Surface Features: It calculates descriptors critical for solubility, like the protein’s surface electrostatic potential and hydrophobicity.
By fusing these three types of information, Pro4S can more accurately determine a protein’s solubility (qualitative classification) and provide a specific solubility value (quantitative regression). This unified approach to handling both qualitative and quantitative tasks is one of the things that sets it apart from previous models.
The researchers tested Pro4S on several public datasets. In qualitative classification tasks, its AUC reached 0.725. In quantitative regression tasks, its R² was 0.558. Both metrics outperform the current top prediction tools in the field.
Pro4S can also guide the design of new proteins. Researchers applied it to de novo designed proteins and found that the model’s predicted solubility scores were highly correlated with experimentally measured protein expression levels. Screening with Pro4S can reduce the proportion of non-expressing proteins by 52.7% while retaining 96.7% of highly expressed ones. This shows the model can efficiently filter out designs that are likely to fail, improving development efficiency.
This work provides new ideas for computational protein engineering. In drug development, whether for designing antibody drugs or developing new enzymes, a reliable solubility prediction tool can save experimental costs and accelerate project timelines.
📜Title: Pro4S: prediction of protein solubility by fusing sequence, structure, and surface 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.11.05.686869v1 💻Code: https://github.com/TEKHOO/Pro4S
2. Which Molecular Representation Do Large Language Models Prefer? InChI and IUPAC Perform Better

In drug discovery, the search for more efficient tools is constant. Large Language Models (LLMs) bring new possibilities, but they also raise a fundamental question: how should we describe a molecule to the model so it can best understand and predict its properties? The traditional method uses the concise SMILES string, but new research suggests this may not be the optimal input for LLMs.
A study examined several common representations in the molecular field: SMILES, the more standardized InChI (International Chemical Identifier), and the IUPAC (International Union of Pure and Applied Chemistry) nomenclature familiar to chemists. Researchers tasked several LLMs, including GPT-4o and Gemini 1.5 Pro, with classic drug discovery tasks, such as predicting whether a molecule can cross the blood-brain barrier or its water solubility.
The results showed that in scenarios with no extra training (zero-shot) or only a few examples (few-shot), InChI and IUPAC often performed better than SMILES.
The researchers analyzed two possible reasons.
First, the structure of InChI and IUPAC names contains more direct information. For example, an InChI string explicitly lists the molecular formula, allowing an LLM to immediately grasp key atomic counts. SMILES is more abstract, requiring the model to expend more computational resources to infer the chemical structure it represents.
Second, the difference is related to the models’ training data. The researchers found that in many biomedical literature databases, IUPAC names appear far more frequently than SMILES. Since LLMs are pre-trained on massive amounts of text data, they are naturally more familiar with IUPAC nomenclature and can process it more efficiently.
This research shows that we can’t just stick to old habits when applying LLMs to chemistry. It’s important to choose a molecular representation that the model can understand better. This is not just about changing the input format, but about thinking how to use the reasoning abilities of these general models to make them interpretable and trustworthy tools in drug discovery. Future research could explore molecular representations even better suited for LLMs, further unlocking their potential in drug design.
📜Title: Molecular String Representation Preferences in Pretrained LLMs: A Comparative Study in Zero- & Few-Shot Molecular Property Prediction 🌐Paper: https://aclanthology.org/2025.emnlp-main.56.pdf
3. AI Drug Discovery: Using ‘Ghost Molecules’ to Fill in Sparse Data
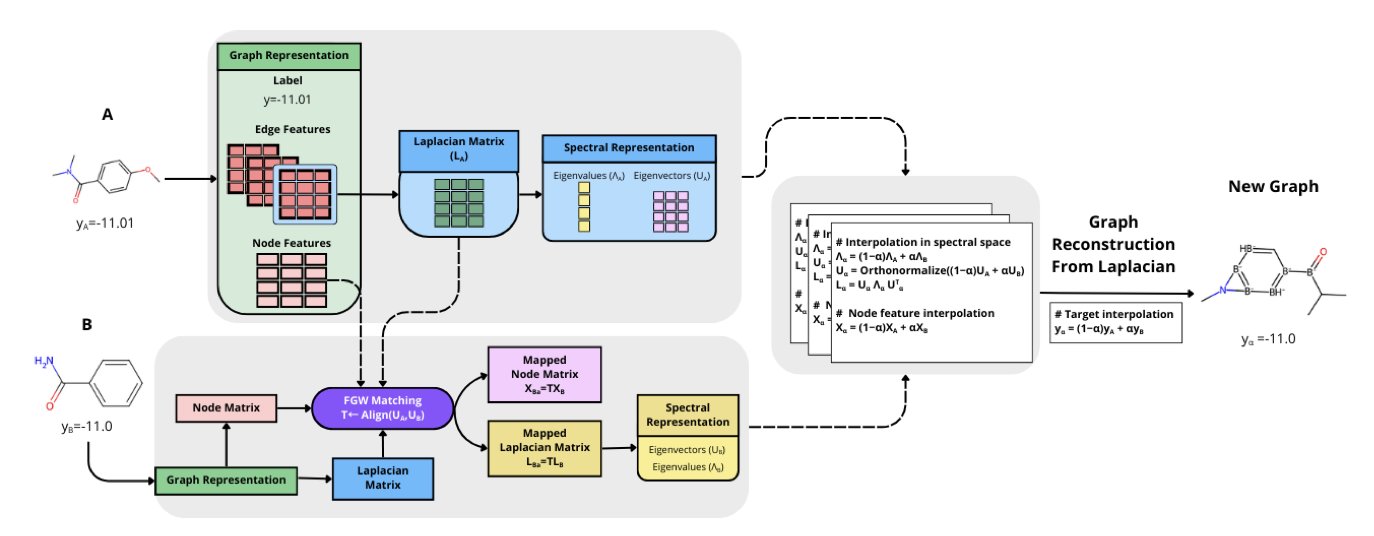
In drug discovery, using computational models to predict molecular properties often runs into the problem of data imbalance. Most of the data is concentrated in a specific range, while data on unique “golden molecules” is scarce. This leads to poor model performance in these critical data regions.
The SPECTRA framework offers a clever solution: create “ghost molecules” to fill the data gaps. These molecules don’t exist in reality, but their chemical structures and properties are perfectly valid.
SPECTRA creates molecules in an abstract mathematical space—the spectral domain. Each molecule can be seen as a graph of atoms (nodes) and chemical bonds (edges). Its structural information can be fully described by the eigenvalues and eigenvectors (the “spectrum”) of its Laplacian matrix.
First, SPECTRA uses a mathematical tool called Gromov–Wasserstein couplings to align two real molecular graphs in the spectral domain, finding their optimal correspondence. Then, by linearly interpolating the Laplacian eigenvalues and node features of these two molecules, it smoothly generates a series of new “spectras.” Finally, it reconstructs the corresponding molecular graphs from these new spectras. This process ensures the generated molecules are chemically valid.
SPECTRA doesn’t generate molecules blindly; it incorporates a “rarity-aware” mechanism. It first uses kernel density estimation to analyze the entire dataset and identify the property ranges with the fewest molecules and the most sparse data. It then focuses on creating “ghost molecules” in these sparse regions, achieving targeted data augmentation.
The researchers validated SPECTRA’s effectiveness on several classic molecular property prediction datasets, such as solubility (ESOL), hydration free energy (FreeSolv), and lipophilicity (Lipo). The results showed that the method significantly improved the model’s prediction accuracy in data-scarce regions without increasing the overall Mean Absolute Error (MAE). Compared to large Transformer models, SPECTRA achieved high predictive performance while running faster, striking a balance between accuracy and efficiency.
SPECTRA provides a precise and efficient data augmentation method. The molecules it generates not only improve model performance but are also created through an interpretable process, helping us understand why the model’s performance improved. This targeted approach to “filling in the data” opens up a new path for solving data imbalance problems in drug discovery.
📜Title: Spectra: Spectral Target-Aware Graph Augmentation for Imbalanced Molecular Property Regression 🌐Paper: https://arxiv.org/abs/2511.04838
4. POEMS: Achieving Interpretable Multi-omics Integration with Sparse Decoding
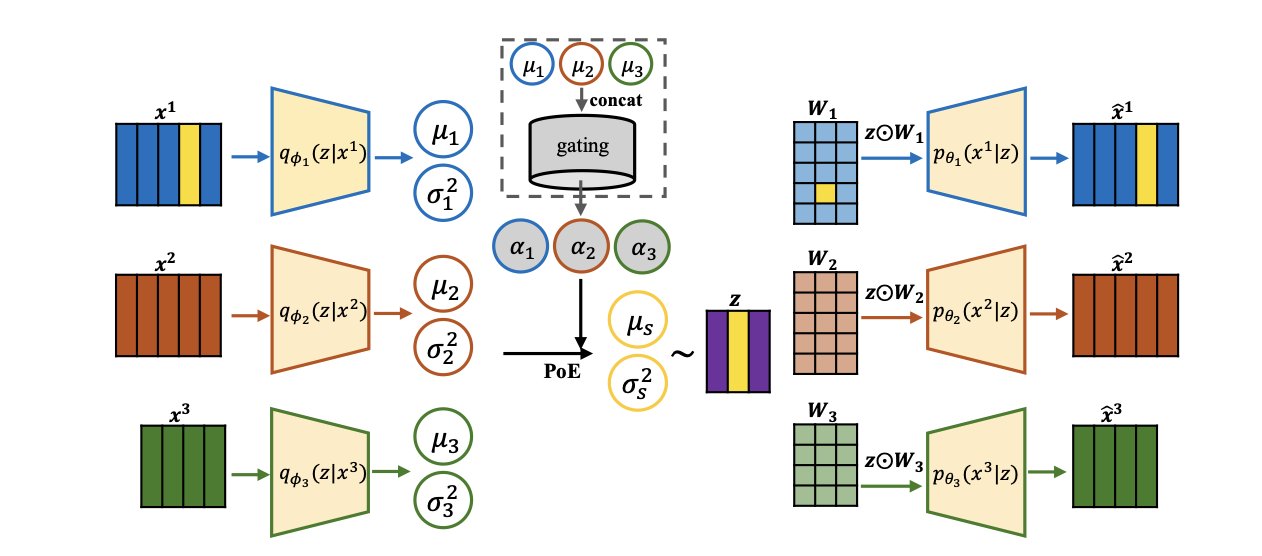
Multi-omics research often faces a dilemma: models are either accurate but hard to interpret, or transparent but lack predictive power. POEMS, a new deep generative framework, offers a balanced solution to this problem.
Its core idea is to build an interpretable latent space. This latent space acts as an information hub, bringing together all different sources of omics data, like genes, proteins, and metabolites. The first design feature of POEMS is sparse decoding. Instead of letting all features into the model, it uses a sparse mapping so that each “factor” in the latent space is associated with only a few of the most relevant omics features. When a factor is activated, we can immediately trace it back to the specific genes or proteins at play, directly identifying biomarkers.
To effectively fuse data from different sources, POEMS uses a Product of Experts (PoE) model. Each omics data source acts as an “expert,” making a judgment based on its own data in the form of a posterior distribution. The PoE’s job is to synthesize these expert opinions to form a more comprehensive consensus. This process not only enhances the model’s understanding of complex biological problems like disease subtypes but also allows us to see clearly how different omics layers work together to paint a full picture of a disease.
POEMS also has a gating mechanism. This mechanism acts like an intelligent mixing board, automatically determining which omics data source is most informative for the current analysis task and assigning it a higher weight. For example, when analyzing a certain type of cancer, if genetic mutations are the primary driver, the genomics data will be given more weight. This ability to dynamically adjust provides a new dimension for understanding the contribution of different molecular layers in disease development.
Researchers validated POEMS on breast and kidney cancer datasets. The results showed that its performance in clustering and classification tasks was on par with current state-of-the-art methods. The latent structures it uncovered were not only biologically plausible but also revealed cross-omics associations and the contributions of each omics layer, which is an important step toward designing more targeted therapies.
POEMS still has room for improvement. Future work could optimize the structure of the latent space and reduce the model’s dependence on hyperparameter settings.
📜Title: POEMS: Product of Experts for Interpretable Multi-omics Integration using Sparse Decoding 🌐Paper: https://arxiv.org/abs/2511.03464v1
5. AI Accurately Predicts RNA-Protein Interactions and Can Even Design RNA in Reverse
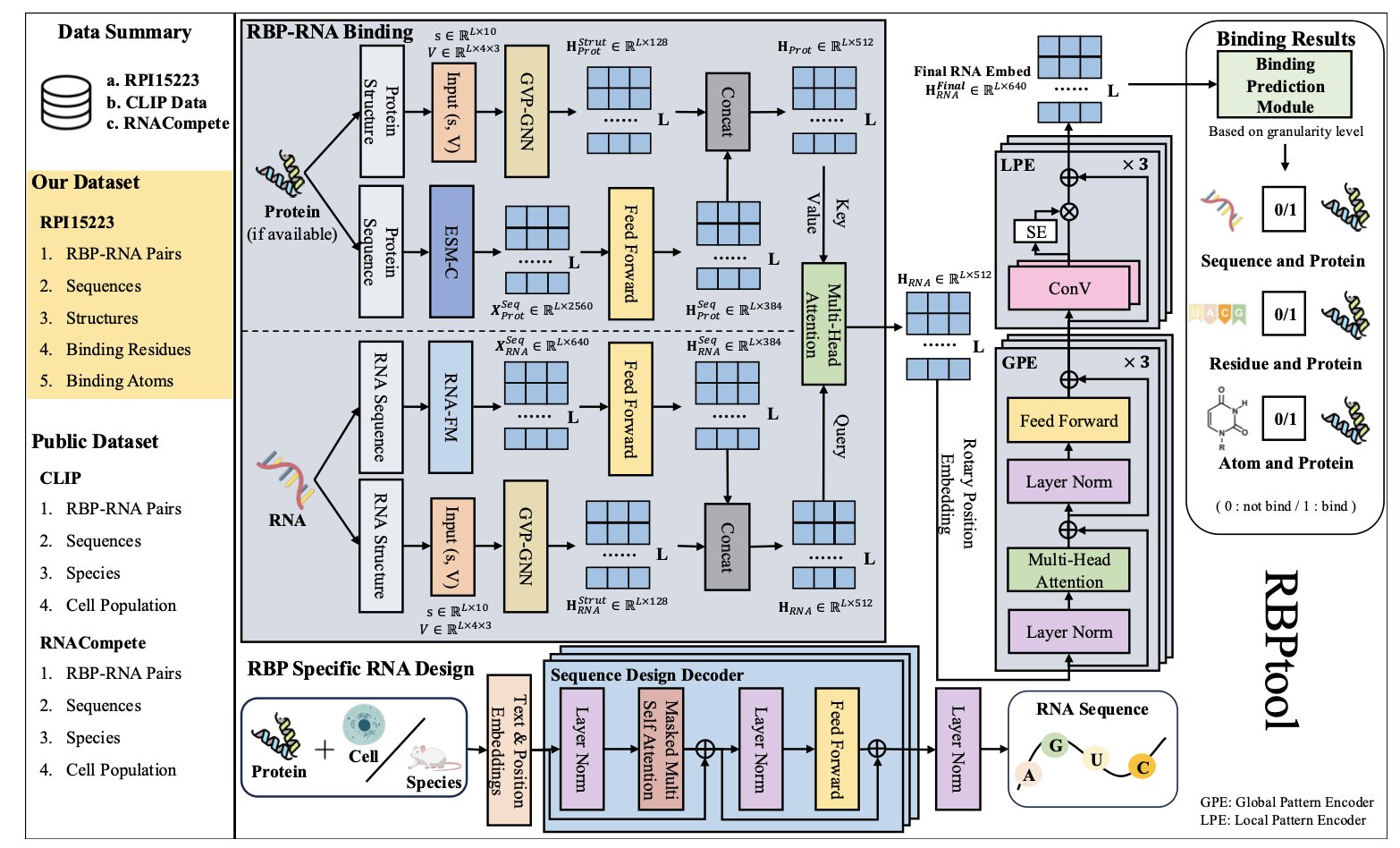
RNA therapeutics are a hot area right now, but designing RNA molecules that can precisely bind to target proteins remains a major challenge. The core problem is accurately predicting how an RNA molecule and its target protein recognize and bind to each other. A new computational framework called RBPtool offers a new approach.
RBPtool’s approach combines two AI technologies: a Large Language Model (LLM) and geometric deep learning.
Predicting RNA-Protein Interactions (RPIs) is like asking an AI to simultaneously read a book (RNA sequence information) and understand a map (RNA 3D structure). Traditional methods often focus on one or the other, resulting in incomplete information.
RBPtool establishes a dual-channel information processing system. The first channel uses an RNA foundation model to “read” the RNA sequence, capturing hidden global patterns. The second channel consists of a geometric vector perceptron module that interprets the RNA molecule’s 3D spatial structure, capturing local geometric features at the atomic and residue levels.
Information from both channels is ultimately combined, allowing RBPtool to make a comprehensive judgment about the binding potential between an RNA and a protein. This strategy of combining global and local information allows it to surpass the accuracy of previous methods.
RBPtool is also an RNA designer. The researchers added an inverse design module to it. A user only needs to specify a target protein and the cell type where it should act, and the model will automatically generate a new RNA sequence that meets the requirements. Experimental validation has shown that these designed RNA sequences perform well in terms of functionality and sequence similarity, opening up a new path for de novo RNA drug design.
The researchers tested RBPtool on several public datasets, including CLIP and RNAcompete, demonstrating its strong performance and generalization ability. An ablation study also confirmed that the large language model and the global pattern encoder are key to its high performance.
RBPtool understands RNA sequences as a language while also considering their complex 3D structures. This allows it to achieve accurate prediction and intelligent design at multiple levels of precision, providing a powerful computational tool for developing effective and precise RNA therapies.
📜Title: RBPtool: A Deep Language Model Framework for Multi-Resolution RBP-RNA Binding Prediction and RNA Molecule Design 🌐Paper: https://aclanthology.org/2025.emnlp-main.110.pdf