
Table of Contents
- Researchers developed a method using sparse autoencoders to make foundation models for single-cell RNA sequencing easier to understand, revealing the biological knowledge the models learn.
- GeoFlow-V3 uses AI to directly generate high-affinity antibodies with a 15.5% hit rate, shortening a screening process that took months to just a few weeks.
- DomDiff applies diffusion models to identify protein domains. By generating labels from noise, it improves the accuracy of boundary prediction and family classification.
- StoPred combines a protein language model with a graph attention network to predict the stoichiometry of protein complexes directly from sequence or structure, outperforming existing methods in both accuracy and efficiency.
- 3D-GSRD designs a clever selective re-masking mechanism that stops a model from “cheating” with 2D graphs, forcing it to truly learn a molecule’s 3D structure.
1. A New Way to Interpret Single-Cell Foundation Models: Sparse Autoencoders Reveal Biological Concepts
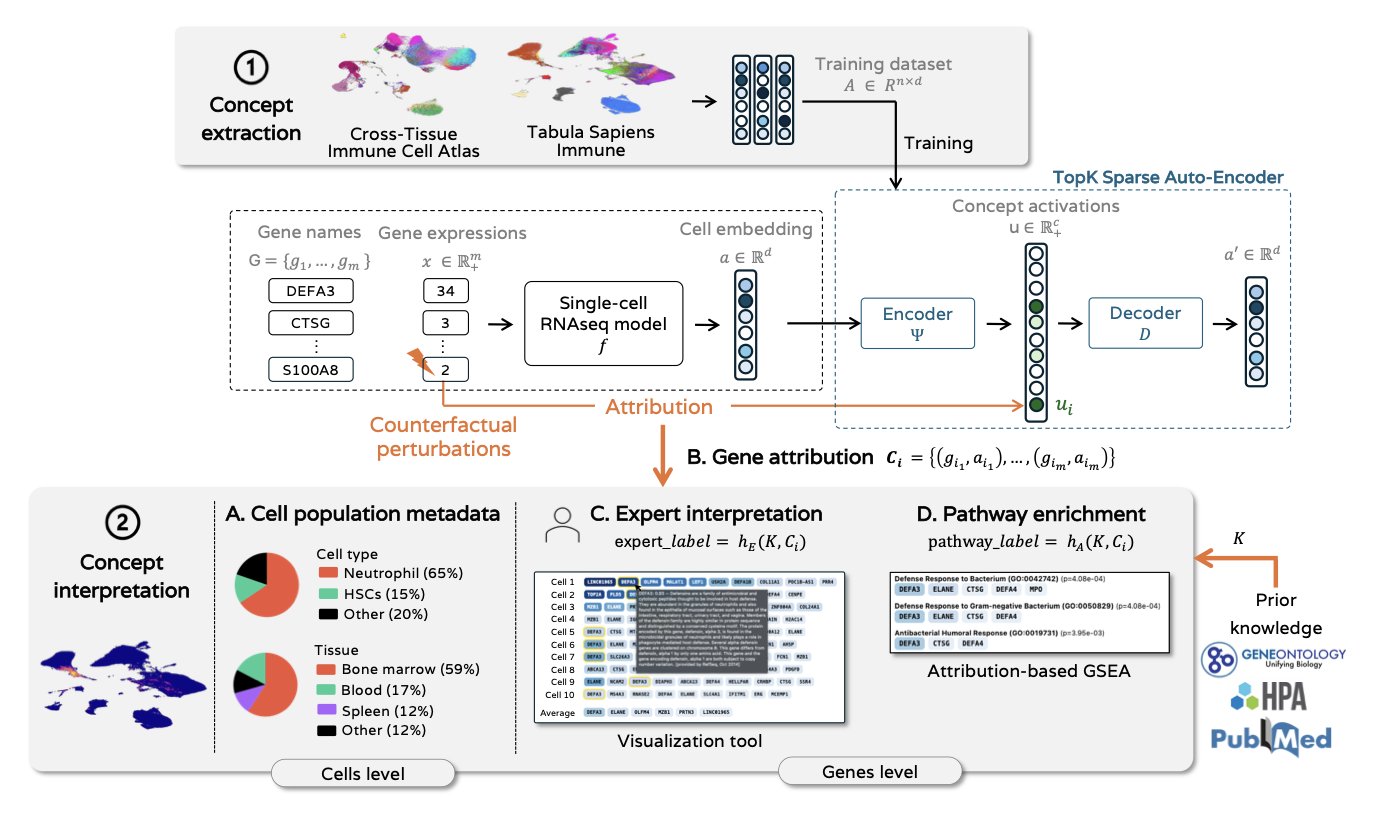
When using deep learning models to analyze single-cell sequencing data, we often face a “black box problem.” The models make good predictions, but how they work is unclear. This is a challenge for scientists who want to use these models to discover new biology. A new study offers a solution.
The core of the research is a sparse autoencoder. It translates the complex neural activity inside a single-cell foundation model into a set of understandable “biological concepts.” Each “concept” might correspond to a group of functionally similar genes, like those involved in a specific metabolic pathway or those that determine a cell’s identity.
Compared to analyzing single neurons, these “concepts” are clearer and more aligned with biology. The study showed that the extracted concepts strongly correlate with known biological signals like cell types and biological processes, making the model much easier to interpret.
To figure out which genes drive each concept, the researchers used counterfactual perturbation attribution analysis. Traditional differential gene expression analysis only shows correlation, but this method can infer causation. By computationally simulating the “knockout” of specific genes and observing which “concept’s” activation is most directly affected, researchers can pinpoint the core genes driving each concept.
This interpretability framework does not hurt model performance. Tests showed that using these “concepts” in downstream tasks like cell classification and cell cycle determination works just as well as using the model’s entire latent space. This means we can gain valuable interpretability without losing prediction accuracy. The method makes the model transparent, opening a new path for using AI tools to explore the mysteries of biology.
📜Title: Discovering Interpretable Biological Concepts in Single-Cell RNA-Seq Foundation Models 🌐Paper: https://arxiv.org/abs/2510.25807
2. GeoFlow-V3: AI Designs Antibodies From Scratch with a 15.5% Hit Rate
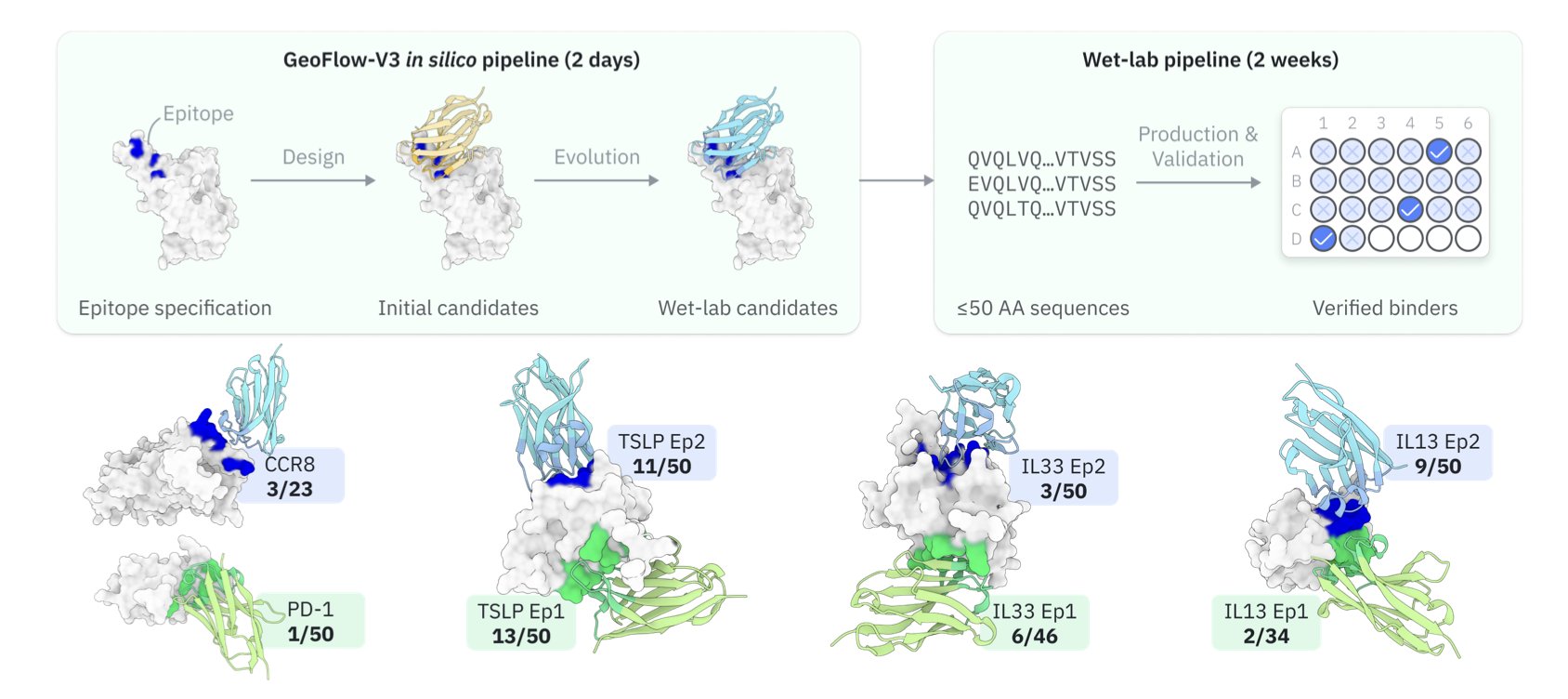
Discovering an antibody is like searching a huge library for one specific key that fits a lock. Traditional methods like phage display are like “brute force” approaches, requiring the screening of massive numbers of molecules, which takes a lot of time and effort. The field has long hoped to use computational methods to directly “design” antibodies, but past designs had very low success rates in experiments.
GeoFlow-V3 tightly integrates generation with structural validation. Think of it as an architect (the generative model) working with a structural engineer (the complex modeler). The architect sketches a design, and the engineer immediately analyzes its structural feasibility. This real-time feedback ensures that the generated antibody sequences are structurally sound and stable in 3D.
GeoFlow-V3 also introduces an “in silico evolution” strategy. It mimics the process of directed evolution: the model generates an initial batch of antibody sequences, evaluates their binding potential to a target with a confidence score, then makes small “mutations” to high-scoring sequences and re-evaluates them. Better results are kept, and the cycle repeats. This process efficiently searches the vast sequence-structure space, pushing candidate molecules toward high affinity.
The research team ran experiments on 8 different therapeutic targets, including difficult ones like cytokines, GPCRs, and immune checkpoints. The average hit rate was 15.5%. That means for every 7 AI-designed molecules tested, one was likely to bind the target. In contrast, previous computational methods might require testing hundreds or thousands of molecules to get a single success. This is an improvement of two orders of magnitude.
For some targets, molecules with nanomolar affinity were found after just one round of wet-lab experiments. The discovery cycle was shortened from months to weeks. The AI provides a small batch of high-probability candidates, avoiding a needle-in-a-haystack search.
This work moves drug discovery from “AI-assisted” to “AI-driven,” where AI is a creator, not just a screening tool. The framework is highly extensible and can be used to design bispecific antibodies, peptides, macrocycles, and even enzymes, opening new possibilities for biologic drug development.
📜Title: Rapid De Novo Antibody Design with GeoFlow-V3 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.20.682964v1
3. DomDiff: A New AI Tool for Defining Protein Domains
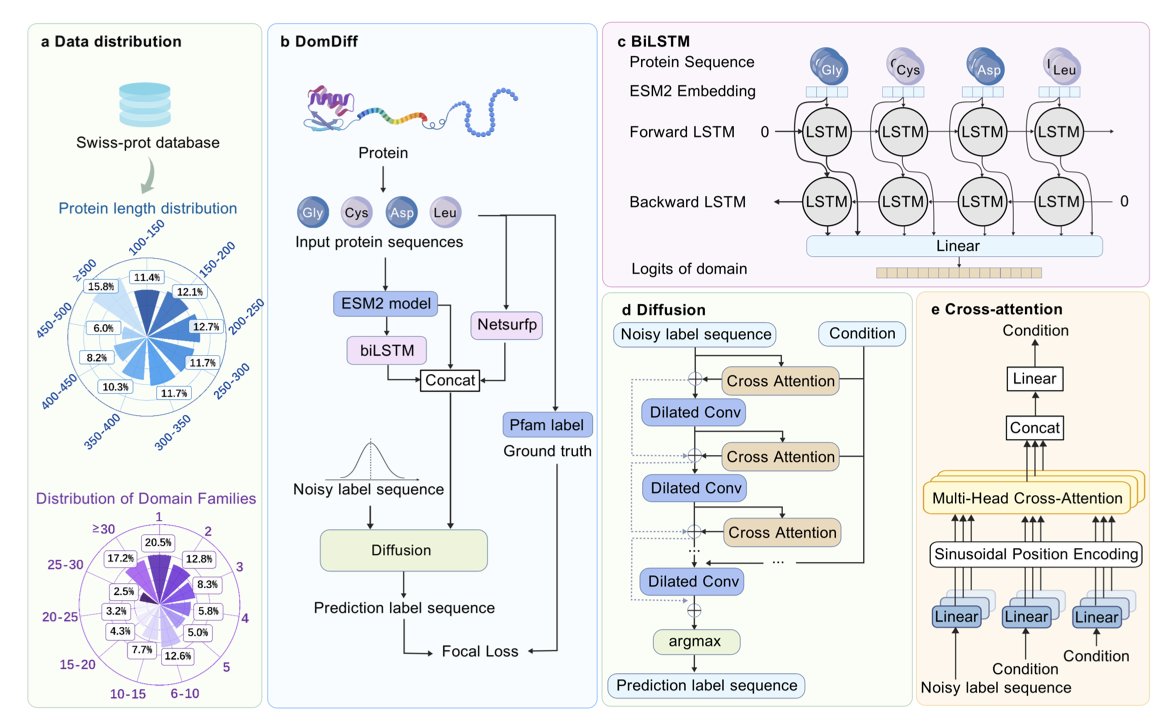
To understand how a protein works, you first need to identify the boundaries of its domains. This is like figuring out the chapters of a book before you can understand the story. Traditional methods often struggle with fuzzy boundaries or rare protein families. DomDiff offers a completely new way to solve this.
DomDiff reframes domain identification as a “generation” task.
Imagine a screen full of static (Gaussian noise), and a model that can gradually turn that noise into a clear image. That’s the idea behind a diffusion model. DomDiff uses this approach, starting with a random set of domain labels and iteratively “denoising” them to produce an accurate domain map.
Here is how the process works:
First, the model takes a protein sequence and uses embeddings from ESM2 (Evolutionary Scale Modeling 2), a powerful large language model for proteins. These ESM2 embeddings provide rich contextual information for each amino acid.
Next, the model incorporates secondary structure information predicted by NetSurfP (like α-helices and β-sheets) and prior knowledge from a bidirectional long short-term memory network (biLSTM). This allows the model to understand not just the amino acids but also the protein’s local structural patterns.
At each denoising step, DomDiff uses this information to correct the random labels. Early steps fix large-scale partitioning errors, while later steps fine-tune the boundaries down to a single amino acid.
This generative approach is better at handling ambiguous boundary regions because it deals with uncertainty through gradual optimization.
It also performs well on rare protein families. DomDiff learns the underlying rules for generating domains, so it can generalize better to “few-shot” cases where data is sparse. This is critical for annotating new genomes.
On public datasets, DomDiff’s boundary detection accuracy is 12.6% higher than the previous top model, and its classification accuracy is 4.2% higher.
More accurate domain annotation leads to a clearer understanding of function. When designing a molecule to inhibit or activate a protein, a precise map of catalytic and regulatory domains makes drug design more targeted and efficient.
The next step is to integrate 3D structural information. If today’s DomDiff is drawing an accurate “floor plan” of a protein, adding 3D structure will allow it to generate a complete “architectural blueprint.”
📜Title: DomDiff: Protein Family and Domain Annotation via Diffusion Model and ESM2 Embedding 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.28.685005v1 💻Code: https://github.com/zhangchao162/DomDiff.git
4. StoPred: AI Accurately Predicts the ‘Recipe’ for Protein Complexes
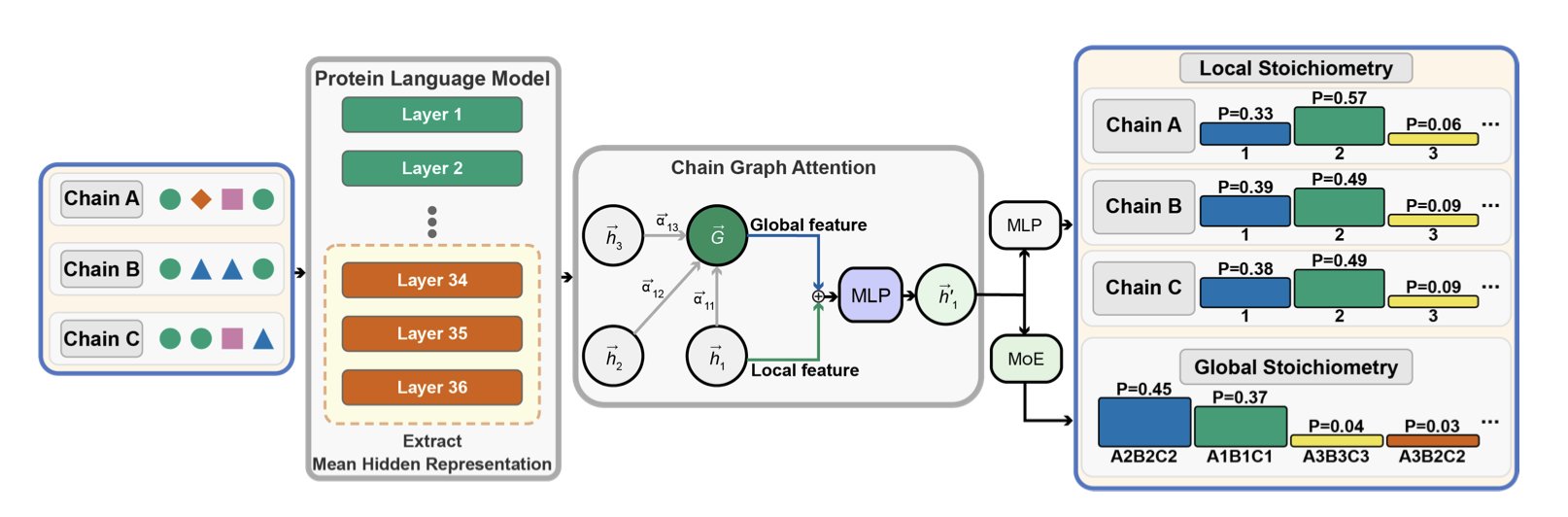
In drug discovery, many protein targets don’t act alone. They form teams, called protein complexes, to carry out biological functions. Figuring out how many members (subunits) are on the team, and how many of each type, is the first step to understanding what they do. This “team recipe” is called stoichiometry. If you get the recipe wrong—for instance, studying an A3B3 complex as if it were A2B2—all subsequent work on structural analysis, functional studies, and drug design could be like “renovating the wrong building.”
Traditional methods for predicting stoichiometry rely on finding known complexes with similar structures to use as templates. But if no template exists for your target protein, prediction becomes difficult.
The StoPred tool offers a new approach. It first uses a protein language model (pLM) to “read” the amino acid sequence of each candidate subunit. The pLM is like an AI that has read countless protein sequences and understands their “grammar,” allowing it to extract deep information about folding and function from the sequence alone.
After getting the details on each potential “team member,” a graph attention network (GAT) decides how to form the team. The GAT acts like a team coach, evaluating all members and simulating their interactions. It focuses on the key interactions that determine the team’s stability and ultimately calculates the most plausible team composition.
The entire process is based purely on sequence or structural features. It needs no templates and no predefined list of possible combinations.
StoPred’s performance is excellent. For homomeric complexes (where all subunits are identical), its Top-1 accuracy is 16% higher than the previous best method. For the more complex and common heteromeric complexes (containing different subunits), this advantage grows to 41%, a significant improvement.
A comparison with AlphaFold3 highlights StoPred’s unique value. AlphaFold3’s scoring systems (like pLDDT) are designed to assess the accuracy of a structural model, not the correctness of its stoichiometry. So, AlphaFold3 might give a high score to a structurally perfect but incorrect complex (like A2B2). StoPred, however, is trained specifically to predict stoichiometry, making it more accurate at identifying the correct recipe (like A3B3).
StoPred runs quickly and can serve as an efficient pre-screening tool. Before committing significant computational resources to modeling with tools like AlphaFold-Multimer, researchers can use StoPred to quickly identify the most likely stoichiometry. This helps avoid wasting time and computing power on incorrect models, making research more efficient.
Future work includes improving structural feature extraction, integrating multiple sources of information (sequence, structure, templates) into a unified model, and extending its application to protein-nucleic acid complexes.
📜Title: StoPred: Accurate Stoichiometry Prediction for Protein Complexes Using Protein Language Models and Graph Attention 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.20.683515v1
5. 3D-GSRD: A New AI Model That Better Understands 3D Molecular Structure
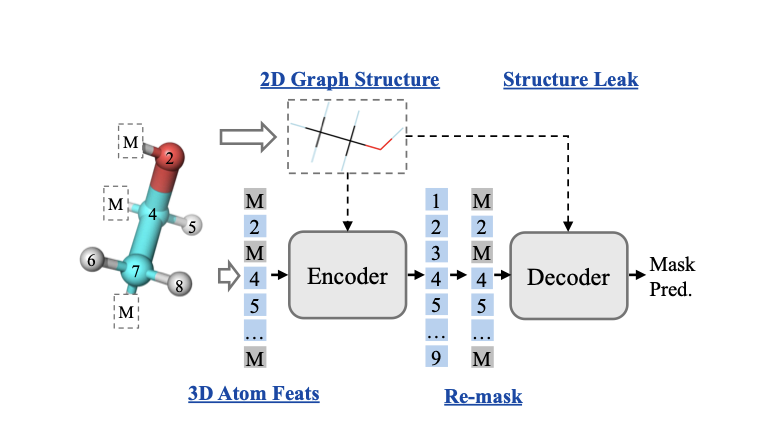
AI-aided drug discovery requires tools that can understand the three-dimensional (3D) structure of molecules. A molecule’s biological activity, like how it binds to a target protein, depends entirely on its 3D conformation. But teaching an AI model to learn 3D information from 1D or 2D data has always been a challenge.
Many self-supervised learning models use a “Masked Graph Modeling” (MGM) strategy. It’s like a fill-in-the-blanks exercise: part of a molecule is hidden, and the model has to predict it. But models often take a “shortcut.” They can guess the hidden atom just by looking at the surrounding 2D chemical bonds, without ever learning the complex 3D spatial arrangement. This “cheating” bypasses the real 3D learning task.
The 3D-GSRD model proposed in this paper is designed to solve this “cheating” problem.
Its core mechanism is “Selective Re-mask Decoding” (SRD). The training happens in two steps. In the encoding phase, the model sees a partially masked molecule. In the decoding phase, the researchers show the decoder the complete 2D molecular graph but again hide the 3D coordinates of the masked part.
This forces the decoder to rely entirely on the encoder’s output to reconstruct the atom’s 3D coordinates, because it can no longer find shortcuts in the 2D structure. The encoder is forced to compress key 3D geometric information into its representation of the molecule. It’s like an engineer who can’t just place a part based on its location on a blueprint, but must also understand its precise orientation and function within the overall 3D structure.
The entire architecture was carefully designed to achieve this goal.
The new method’s effectiveness was validated on the MD17 benchmark. MD17 specifically evaluates a model’s ability to predict 3D properties like molecular energy and forces. Test results showed that 3D-GSRD achieved the best scores on 7 out of 8 tasks, proving that this method of forcing the model to learn 3D information works.
The authors also noted the model’s limitations. For example, the pre-training dataset is relatively small. The model’s potential could be further unlocked by using larger datasets like PubChemQC. In the future, this pre-training paradigm could be used not just for property prediction, but also for 3D molecular generation—a more exciting prospect for drug design.
📜Title: 3D-GSRD: 3D Molecular Graph Auto-Encoder with Selective Re-mask Decoding 🌐Paper: https://arxiv.org/abs/2510.16780v1 💻Code: https://github.com/WuChang0124/3D-GSRD