
Table of Contents
- The Omni-DNA model combines Natural Language Processing with DNA sequence analysis, enabling it to “read” ultra-long genomes and “write” their functional annotations, opening new paths in genomics research.
- The RxnCaption framework combines molecule detection with a Large Vision-Language Model, transforming the complex task of parsing chemical reaction diagrams into a simpler image captioning problem, improving accuracy and generalization.
- The S2VM framework treats drug molecules as images and uses self-supervised learning to integrate their structural information, providing a more accurate and interpretable method for predicting drug-drug interactions.
- By combining deep learning with conformational sampling, DeepAAAssembly surpasses AlphaFold3 in the accuracy and reliability of predicting antibody-antigen complex structures.
- The new PDDMA-DTI framework borrows from physics diffusion principles to more accurately and reliably predict drug-target interactions by simulating how information spreads through a network.
1. Omni-DNA: AI Reads DNA, Unlocking the Long Narrative of the Genome
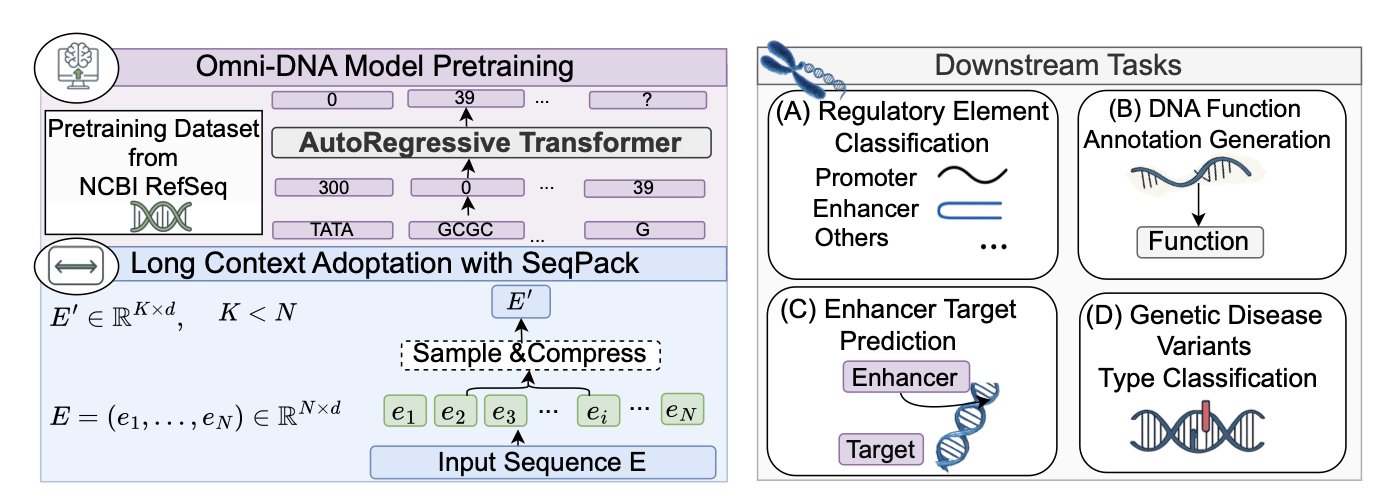
In drug development, we often deal with various targets and sequences. This paper on Omni-DNA marks another step forward for AI in genomics: teaching a machine to understand DNA just like it understands human language.
A New Way to “Compress” DNA Sequences
Analyzing ultra-long DNA sequences used to be a tough problem. It was like reading a massive novel without punctuation or chapters—easy to get lost, and computationally expensive. Previous models, such as the Nucleotide Transformer, still struggled with long sequences.
The developers of Omni-DNA created a compression method called SEQPACK. It efficiently “packages” extremely long gene sequences (over 450,000 base pairs) without losing key information. This allows the model to process longer sequences with fewer computational resources and capture interactions between distant gene-regulatory elements, like an enhancer and its target gene. This is critical for understanding complex gene-regulatory networks.
When DNA Sequences Meet Natural Language
The highlight of Omni-DNA is that it truly connects DNA with human language. The researchers treat DNA not just as a string of A, T, C, and G, but as a language with its own grammar and meaning.
They built the SEQ2FUNC dataset to serve as a “translation dictionary” for DNA, mapping DNA segments to functional text descriptions. Using this dataset, Omni-DNA can learn to “translate” DNA sequences. If you input a DNA segment, it can not only identify its type (like whether it’s an enhancer) but also generate a piece of text describing its potential function.
This capability upgrades the model from answering “multiple-choice questions” (classification) to handling “open-ended questions” (generation). It can extract biological meaning from a sequence and express it in human language. This is immensely valuable for functional genomics research. For example, when a new gene mutation is discovered, the model could predict and generate text describing how the mutation might affect gene function, guiding subsequent experiments.
The Power of Multi-Task Learning
The model also uses a multi-task learning strategy. The researchers fine-tuned Omni-DNA on several related biological tasks simultaneously, such as predicting regulatory elements and enhancer-target gene interactions at the same time. Just as a student studying both algebra and geometry can find that the knowledge reinforces itself, this approach helps the model capture common biological signals underlying different tasks. This improves its performance and generalization ability across the board.
From a research scientist’s perspective, Omni-DNA is a higher-performing model and represents a new research paradigm. It deeply integrates mature Natural Language Processing techniques into genomic analysis, bringing us one step closer to “reading the book of life.”
📜Title: Omni-DNA: A Genomic Model Supporting Sequence Understanding, Long-context, and Textual Annotation 🌐Paper: https://openreview.net/pdf/e038f294ab29cc597ce4e261209e5e765c2069f9.pdf
2. A New AI Paradigm: Parsing Chemical Reactions Like Image Captioning
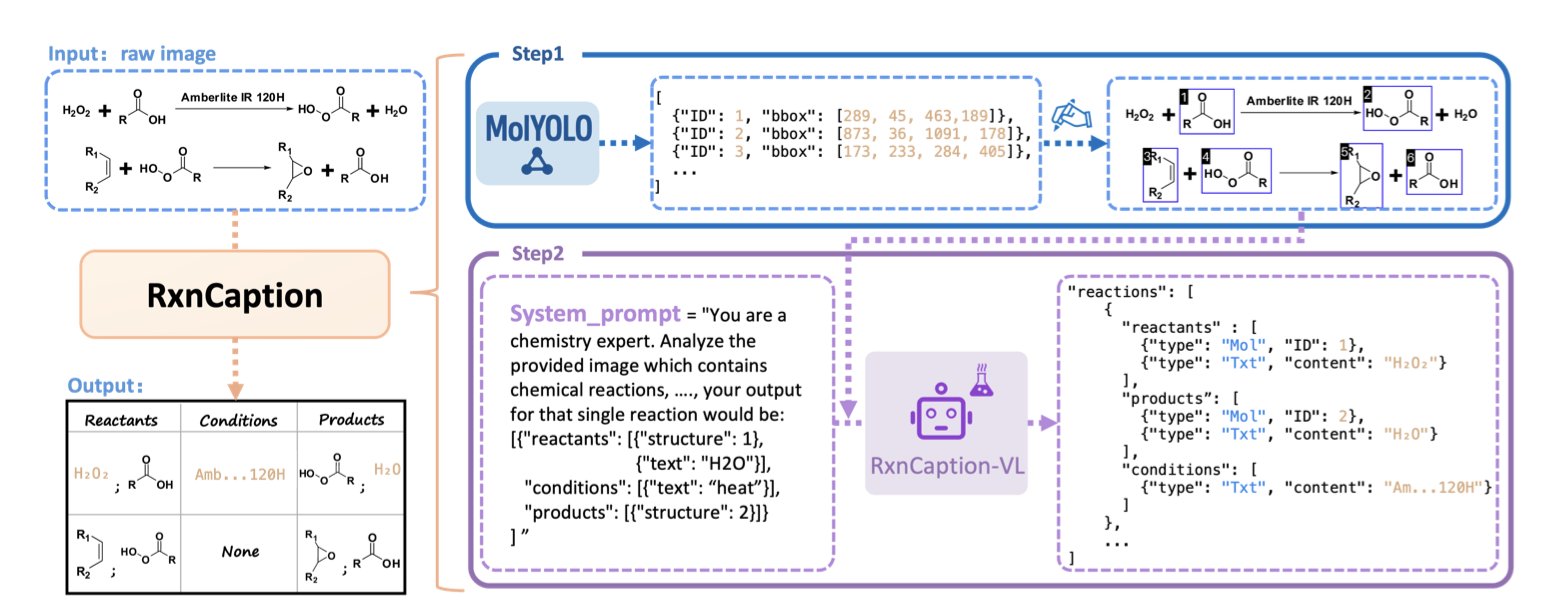
Automatically extracting reaction equations from chemical literature is a persistent challenge in chemoinformatics. Traditional methods require AI models to perform two tasks at once: locate each molecule in a complex reaction diagram and identify its role, such as reactant or product. This is difficult for AI. Even Large Vision-Language Models (LVLMs) are better at understanding image content than performing precise localization.
A Clever Solution
Researchers at Peking University have proposed a new method called RxnCaption. Its core idea is to let the LVLM focus on what it does best.
It works in two steps: 1. Locate Molecules: First, an object detection model specialized in chemical structures, MolYOLO, is trained. This model quickly and accurately draws bounding boxes around all molecules in a reaction diagram and numbers them. 2. Describe Relationships: Then, this annotated image is given to an LVLM. The task becomes much simpler. The LVLM just needs to look at the image and describe the reaction process in natural language, for example: “Number 1 and number 2 are reactants, the arrow points to number 3, so number 3 is the product.”
This “detect first, then describe” strategy transforms a complex visual localization task into an image description task, which LVLMs are good at.
A Larger Dataset and More Realistic Challenges
To validate their method, the researchers built a new dataset, RxnCaption-11k. It was extracted directly from published chemistry papers, making it an order of magnitude larger than previous datasets and including a wide variety of complex layouts.
Older datasets had neat layouts. In contrast, the reaction diagrams in RxnCaption-11k have diverse arrangements—horizontal, vertical, or even circular—which better tests a model’s ability to generalize.
Experimental results show that the RxnCaption model outperformed the previous state-of-the-art model (RxnScribe) on all metrics. Its advantage was even more pronounced on the new, more challenging dataset, proving the method’s accuracy and ability to handle complex, real-world scenarios.
Future Directions
This method currently relies mainly on visual information. The researchers believe that future work should involve integrating chemical knowledge more deeply into the model. If the AI could understand the chemical principles behind a reaction, such as identifying reaction centers, the parsing accuracy would improve further.
📜Title: RxnCaption: Reformulating Reaction Diagram Parsing as Visual Prompt Guided Captioning 🌐Paper: https://arxiv.org/abs/2511.02384 💻Code: https://github.com/songjhPKU/RxnCaption
3. AI Predicts Drug Interactions: A Look at the New S2VM Framework
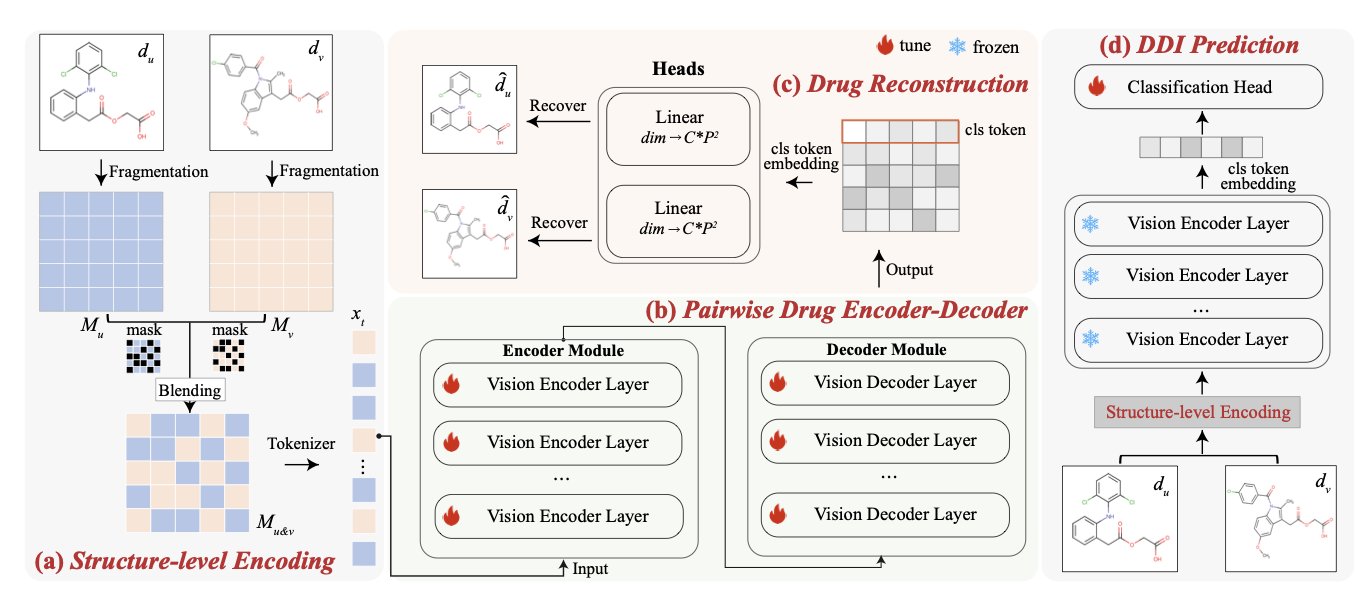
Predicting the effects of taking two drugs together, known as Drug-Drug Interactions (DDIs), is a core challenge in drug development and clinical practice. Incorrect combinations can reduce a drug’s effectiveness or even cause severe toxic side effects. Traditional methods rely on expensive, time-consuming lab screening or computational models with limited accuracy and generalization. A new framework called S2VM offers a clever solution.
Unlike conventional methods that represent drugs using SMILES strings or molecular graphs, S2VM starts by rendering each drug molecule into a 2D image. It’s like taking a “passport photo” of the molecule, which visually preserves its spatial conformation and the relative positions of its functional groups.
Its core step is “Visual Blending.” Instead of processing the two drug molecules separately and then combining the information, S2VM randomly takes patches from the two molecular “photos” and blends them into a single new input image, like a puzzle.
This approach forces the model, when processing this “blended image,” to pay attention to both the internal structure of each molecule (like specific rings or functional groups) and the new spatial relationships that might form between them. It’s like cutting up and reassembling photos of two people, which prompts an observer to notice subtle features that might create a connection.
S2VM uses Self-supervised Learning for pre-training, so it doesn’t need large amounts of labeled DDI data. It learns the structural language of drugs on its own by “looking” at massive numbers of unlabeled drug molecule pairs in a “puzzle-and-restore” type of task. For instance, the model might have to predict the original placement or ownership of shuffled patches from a “blended image.”
Through this pre-training, the model learns to understand the structure of individual molecules from a visual perspective and the patterns that can emerge when different molecular structures are combined. Afterward, it only needs to be fine-tuned with a small amount of known DDI data to accurately predict the interaction between any two drugs.
On several mainstream DDI prediction benchmark datasets, S2VM outperformed most existing methods. Its advantage is particularly clear when dealing with sparse data or new drugs. Self-supervised learning gives it stronger generalization capabilities, making it effective even when faced with drug combinations it has never seen before.
The model also has a degree of interpretability. Because it’s image-based, we can use visualization techniques to see which parts of the molecules the model focuses on during prediction. For example, the model might highlight a hydroxyl group on one molecule and a carboxyl group on another, suggesting a potential hydrogen bond that could cause an interaction. This helps us understand the mechanism of DDIs and can guide future drug design.
Although treating molecules as images doesn’t perfectly capture 3D spatial information, S2VM presents a new approach and provides an efficient, intuitive tool for solving the DDI prediction problem.
📜Title: Self-supervised Blending Structural Context of Visual Molecules for Robust Drug Interaction Prediction 🌐Paper: https://openreview.net/pdf/8c8be03ed80289c37107d98f38f490b948870f50.pdf
4. DeepAAAssembly: AI Predicts Antibody-Antigen Structures, Surpassing AlphaFold3
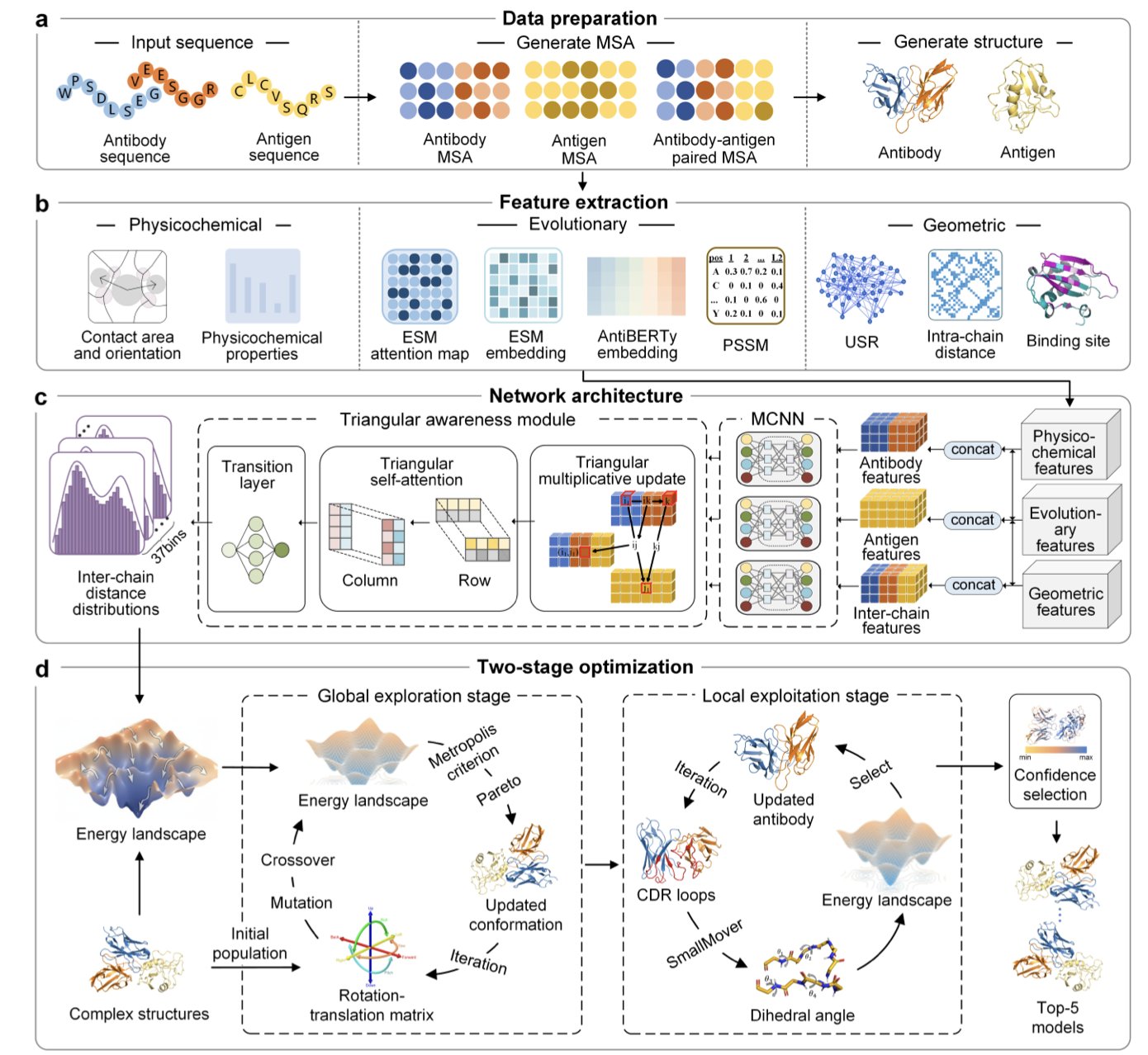
Predicting how an antibody binds to its antigen is a tricky problem. An antibody’s Complementarity-Determining Region (CDR) loops are flexible like soft noodles, making them difficult for traditional physics-based docking methods to handle. While AI models like AlphaFold2/3 have shown promise, their performance on protein-protein interactions (PPI) can be inconsistent, especially with highly flexible systems like antibodies.
Researchers have developed a new method, DeepAAAssembly, that combines the strengths of both approaches.
Its core idea is this: first, use deep learning to predict which amino acid residues on the antibody and antigen will come close to each other. This gives you a “contact map.” These distance predictions are then converted into an energy function that guides the subsequent structural search.
Next comes a two-step conformational sampling process:
- Global Docking: The program roughly explores various possible orientations of the antibody relative to the antigen, quickly identifying a few promising binding poses.
- Local Refinement: Once the general orientation is set, the program finely adjusts the conformation of the highly flexible CDR loops to ensure they fit perfectly against the antigen’s surface.
DeepAAAssembly was compared to AlphaFold3 on a test set of 67 antibody-antigen complexes with known structures.
The results showed that DeepAAAssembly’s average DockQ score was 12.9% higher than AlphaFold3’s. DockQ is the gold standard for evaluating the quality of protein docking models; a higher score means the model is closer to the real structure. DeepAAAssembly successfully upgraded many models predicted as “incorrect” by AlphaFold3 to “acceptable” or “medium” quality, showing it is more reliable for difficult cases.
The method also has a built-in screening mechanism that can automatically select the best model—the one that is most structurally sound and energetically stable—from the multiple structures it generates.
DeepAAAssembly cleverly combines the pattern recognition ability of deep learning with the physical sampling strengths of traditional docking methods. The AI provides high-level “intelligence” (residue distances), while conformational sampling performs detailed “on-site exploration” under the constraints of physical rules. This hybrid strategy offers a powerful framework for solving complex and flexible interaction systems like antibody-antigen binding.
📜Title: High-accuracy structure modeling for antibody-antigen complexes 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.11.03.686275v1
5. Physics-Inspired Diffusion Model Predicts Drug Targets with Higher Accuracy
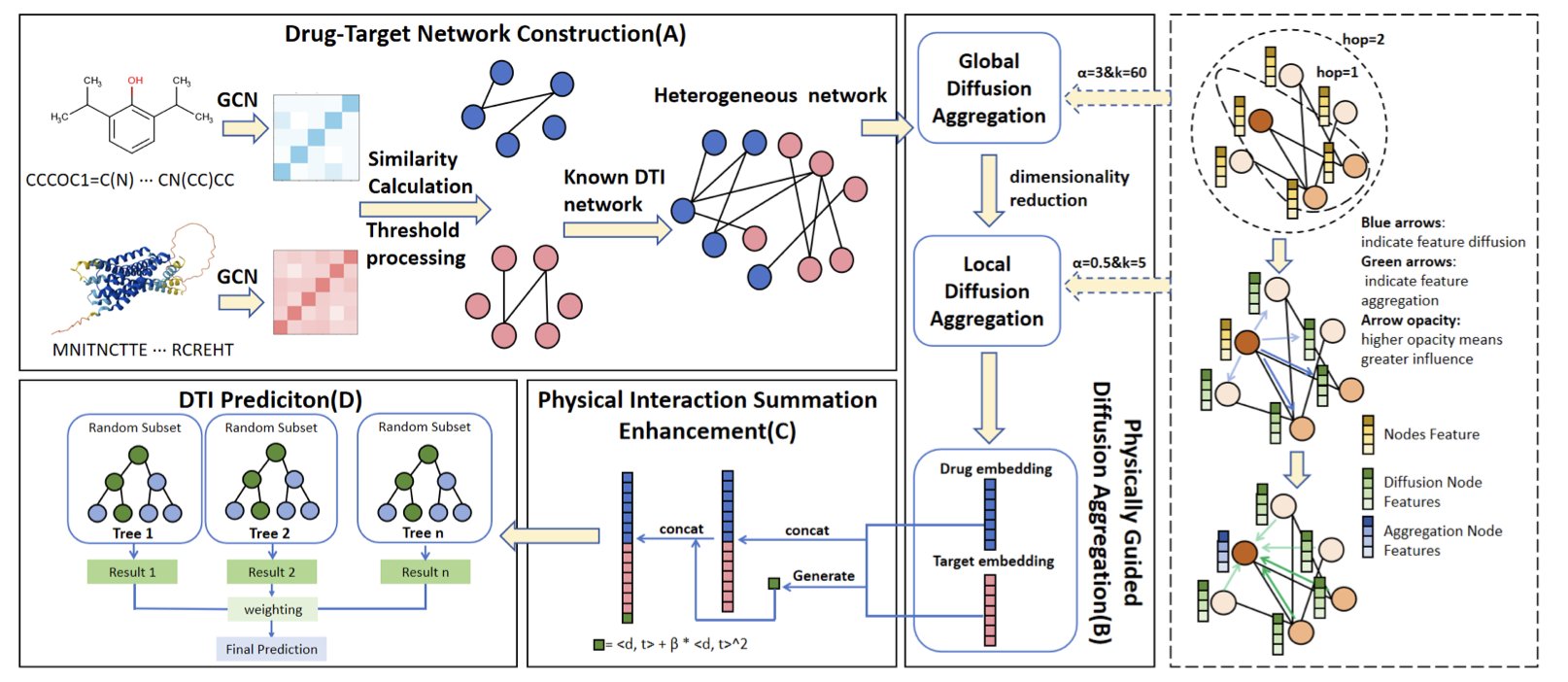
Predicting which protein target a drug will bind to in the body is a central question in drug discovery. Graph Neural Networks (GNNs) often struggle with this problem because it’s hard for them to capture both global information and local features at the same time. Too few message-passing layers give the model a limited view; too many, and node features tend to become too similar, leading to the “over-smoothing” problem where they lose their uniqueness.
To solve this, researchers took inspiration from physics and developed the PDDMA-DTI framework. Its core idea mimics the process of ink diffusing in clear water.
The framework operates in two steps:
First, it uses a two-layer diffusion scheme. The first layer is “global diffusion,” which uses the graph Laplacian to simulate the long-range spread of information across the entire drug-target network. This is like a drop of ink spreading out in water, allowing information to travel across different types of nodes, such as drugs and targets.
The second layer is “local diffusion,” which smooths information within a node’s immediate neighborhood. This is like how ink, as it spreads, first evenly colors a small area around it. To ensure this process is stable and controllable, the researchers used an implicit Euler method to avoid numerical issues.
The framework’s most clever feature is its adaptive stopping rule. The model monitors whether the information at each node has reached a stable “steady-state.” Once it does, the diffusion stops. This ensures that global information has flowed sufficiently while preventing local features from becoming blurred by too much mixing, striking a good balance.
Finally, to make the model better at identifying true drug-target interactions, the researchers added a “Physical Interaction Summation Enhancement” module. This module uses an energy model with both linear and quadratic terms to specifically amplify the signal of highly correlated drug-target pairs. It’s like shining a spotlight on two closely related people in a crowd to make them stand out.
Tests on several standard datasets showed that PDDMA-DTI is more accurate and robust than current leading methods. Its advantage is particularly noticeable in scenarios with imbalanced data. This is crucial for real-world drug discovery, where the vast majority of drugs and targets do not interact.
📜Paper: Physics-Diffusion-Driven Multiscale Aggregation for Drug-Target Interaction Prediction 🌐Link: https://www.biorxiv.org/content/10.1101/2025.11.03.686185v2