
Table of Contents
- IRIS is a machine learning tool for re-ranking poses that significantly improves RNA-ligand docking accuracy by learning from a large set of real structures, solving a long-standing problem in the field.
- The HiF-DTA model achieves high precision in Drug-Target Affinity (DTA) prediction through an innovative hierarchical feature learning network, opening a new path for computational drug discovery.
- The ProAffinity++ algorithm combines protein sequence and structural information to accurately predict protein-protein binding affinity using Graph Neural Networks, outperforming current state-of-the-art methods.
1. Machine Learning Masters RNA Docking: IRIS Tool Boosts Pose Prediction Accuracy
Molecular docking results can be frustrating, especially for flexible targets like RNA. Computational programs generate many possible binding poses, but it’s hard to tell which one is closest to reality. Traditional scoring functions don’t perform well here because RNA binding pockets are less defined than those in proteins, and the interactions are more complex. Researchers can spend a lot of time chasing what turns out to be a false prediction from an algorithm.
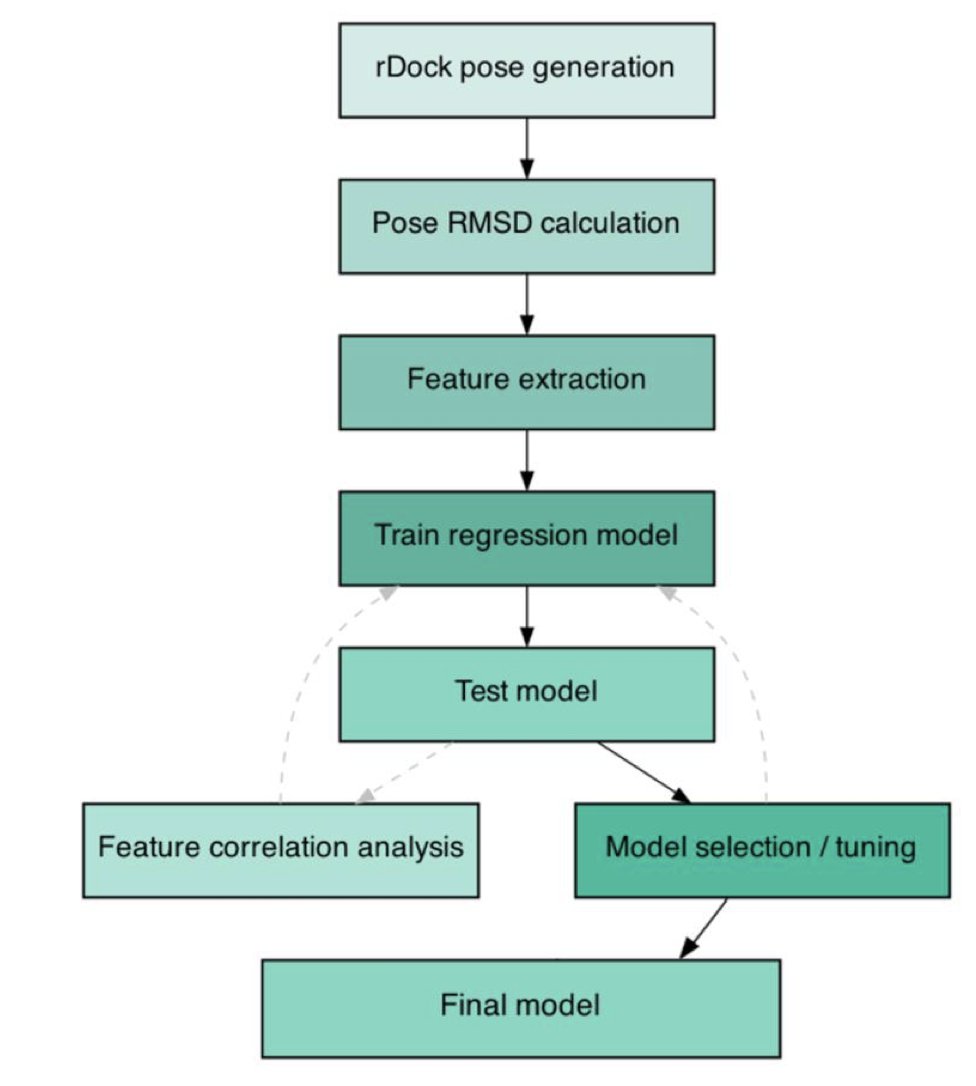
A new tool called IRIS offers a different approach to this problem. It acts like an experienced reviewer. It takes the poses generated by existing docking software, like rDock, and uses a machine learning model to re-rank them and select the most credible results.
What makes the model effective?
First, the data. The model’s success is built on high-quality, large-scale training data. The research team compiled the largest dataset of nucleic acid-ligand complexes to date, containing 1,356 structures. The model learns from these real binding patterns, much like a novice learns by observing a master’s work and gradually develops an intuition for what is good.
Second, the features. The model makes its judgments based on a carefully designed set of features, including:
- rDock scores: The original scores from the docking program.
- AutoDock Vina scores: Scores from another widely used docking tool.
- Ligand-specific features: Properties like the number of rotatable bonds.
- Interaction features: Detailed descriptors of how the ligand and target interact.
This combination of features captures the binding process more completely, including electrostatic interactions, spatial complementarity, and the geometric relationship between the ligand and target. It’s like a detective using multiple pieces of evidence to solve a case, which makes the conclusion more reliable.
How well does it work?
The numbers show its effectiveness. After re-ranking with IRIS, the success rate of finding a near-native pose (RMSD < 2.0 Å) within the top five candidates increased from rDock’s default of 55.4% to 73.2%. This is a 31% relative improvement.
In drug discovery, this increase in accuracy allows computational and medicinal chemists to screen and design molecules with more confidence. When the probability of a computational prediction being correct goes up, they can commit resources to synthesis and testing with less risk of wasting effort on a wrong lead.
A smart design choice was to include both RNA and DNA complexes in the training data. This allowed the model to learn more general principles of nucleic acid-ligand interactions, making it effective for both RNA and DNA targets.
A practical feature of IRIS is its ability to directly predict the Root Mean Square Deviation (RMSD) value for each pose. It quantitatively assesses how far a pose is from the real structure, giving researchers a more refined basis for making decisions.
IRIS is open-source and free, and it can be integrated into existing computational workflows. After running a docking job, processing the results with IRIS provides a more reliable ranking of poses. For researchers in the field of RNA-targeted drugs, this is a practical tool they can use immediately.
📜Title: IRIS: A Machine Learning-Based Pose Re-Ranking Tool for RNA-Ligand Docking 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.24.684232v1
2. HiF-DTA: A New Chapter in Drug-Target Affinity Prediction
Core Idea:
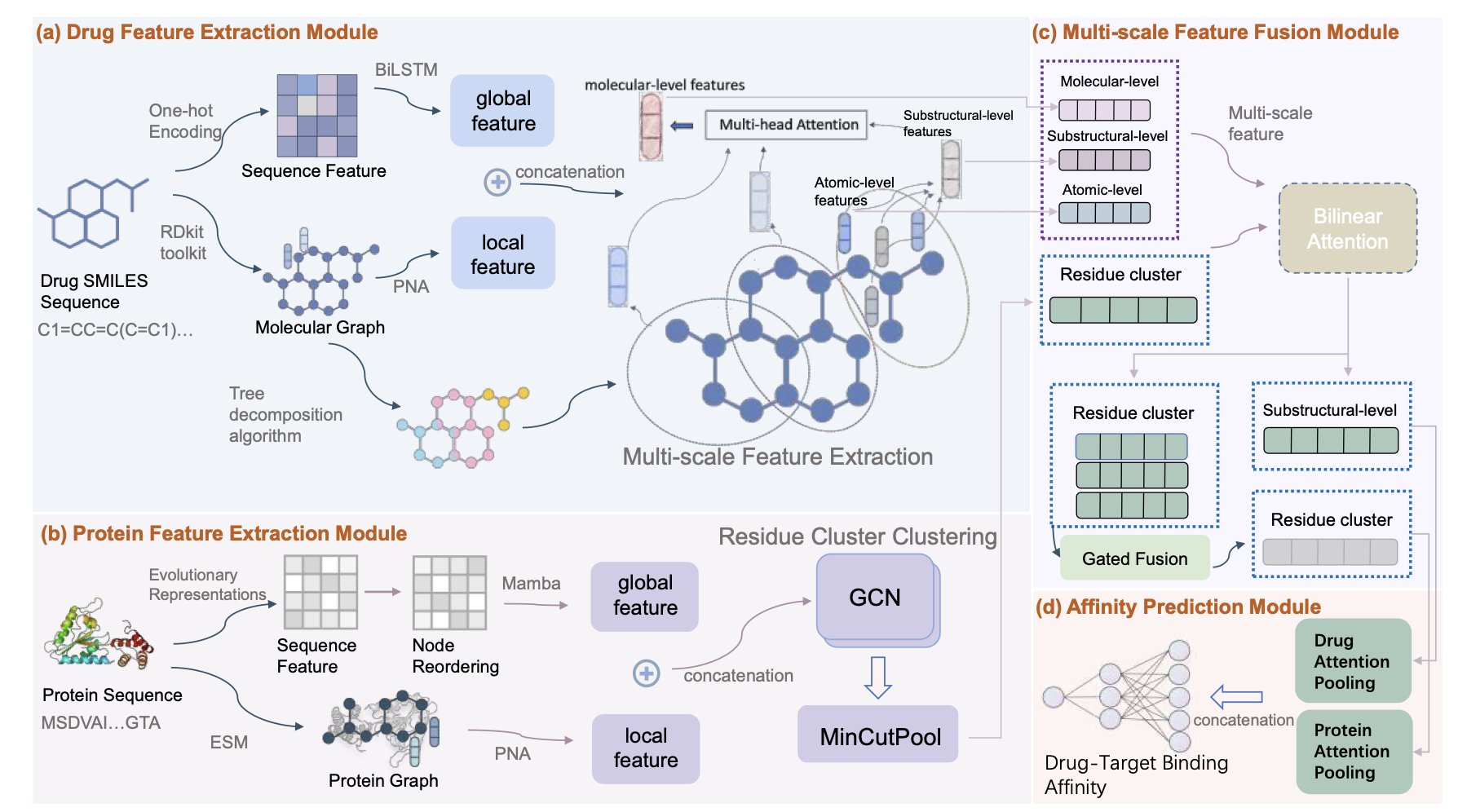
The HiF-DTA model uses a hierarchical network to learn features at multiple scales—from atoms to substructures to the whole molecule—to predict drug-target affinity. This approach has led to better performance on standard benchmarks.
In Detail:
In computer-aided drug discovery, accurately predicting the binding affinity between a drug molecule and a target protein is key to efficiency and cost. A model called HiF-DTA has recently made progress in this area.
The binding of a drug to a target is a complex, multi-scale interaction that happens in three-dimensional space. Traditional models often focus on sequence information and overlook this fact. This is like looking at a painting’s overall composition but ignoring the brushstrokes.
HiF-DTA was designed with a dual-channel network that processes both global and local features. It breaks down the drug molecule into three levels for analysis: atoms, substructures, and the complete molecule. This is like understanding a mechanical watch not just by its appearance, but by studying its internal gears and springs to see how it works.
To combine information from different scales, the model uses a “multi-scale bilinear attention” module. This module acts like a conductor of an orchestra, coordinating features from different levels (atoms, substructures, molecules) to accurately simulate the complex interactions between the drug and its target.
HiF-DTA’s performance was compared against existing models on three established benchmark datasets: Davis, KIBA, and Metz. The results showed that its concordance index (CI, a metric for ranking accuracy) on the Davis dataset exceeded 0.9 for the first time, reaching 0.9026. This demonstrates its improved predictive power.
To confirm that the model’s design was effective, the researchers conducted ablation studies. The results confirmed that both the global-local feature extraction and the multi-scale feature fusion were critical to the model’s performance. This shows that HiF-DTA’s hierarchical feature learning strategy is successful.
📜Paper Title: HiF-DTA: Hierarchical Feature Learning Network for Drug–Target Affinity Prediction 🌐Paper Link: https://arxiv.org/abs/2510.27281
3. ProAffinity++: Predicting Protein Binding Affinity with Graph Neural Networks
In drug development, accurately predicting the binding affinity between proteins is important for designing more effective drugs. The ProAffinity++ algorithm shows excellent capability in this area.
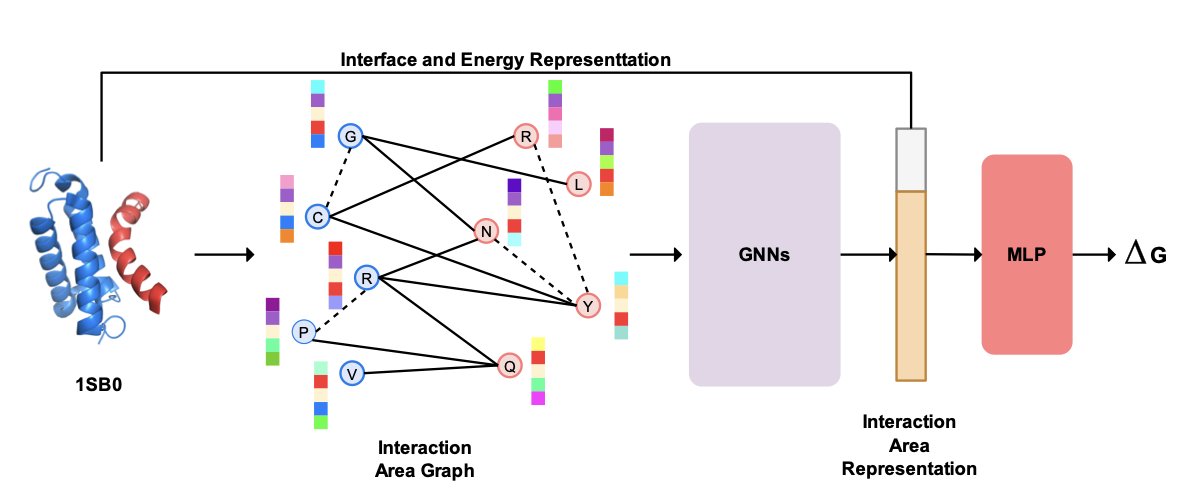
The algorithm’s key idea is that to accurately predict how well two proteins bind, you must consider both their amino acid sequences and their three-dimensional structures. Traditional methods often neglect one of these, leading to a loss of information. ProAffinity++ combines both.
The researchers use graph neural networks to model the protein binding interface as a network, where each amino acid residue is a node. Through a bipartite graph, the algorithm can describe the complex interactions between the two proteins and capture the local microenvironment information that determines binding strength.
To help the model understand protein features, the algorithm incorporates the pre-trained language model ESM-1v and the inverse folding model ESM-IF. ESM-1v extracts biological features from the amino acid sequence, while ESM-IF reveals the deep connections between sequence and structure. Together, they provide rich information for the algorithm.
In tests, ProAffinity++ outperformed existing state-of-the-art methods across the board, including in standard binding affinity prediction and in challenging scenarios like antigen-antibody recognition and missense mutations. This demonstrates that it is a tool with both general applicability and practical value.
The researchers acknowledge that the algorithm can still be improved, for instance, by increasing the depth of the neural network or including more protein conformational data to further enhance prediction accuracy. This opens up new possibilities for computer-aided drug design.
📜Title: ProAffinity++: An End-to-End Algorithm for Predicting Protein-Protein Binding Affinity 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.31.685718v2