
Table of Contents
- SubRec combines a drug’s chemical substructures with a patient’s electronic health records to provide more accurate and safer personalized medication recommendations.
- GeoPep uses the transfer learning capabilities of the ESM3 model, combined with Kolmogorov-Arnold Networks, to efficiently and accurately predict protein-peptide binding sites even without structural data.
- Researchers at Johns Hopkins University developed DL4Proteins, a set of interactive Jupyter Notebook tutorials. Hosted on Google Colab, it gives students and researchers worldwide free access to advanced AI tools for learning and practicing protein structure prediction and design.
1. SubRec: Fusing Drug Substructures and Health Records for Personalized Medicine
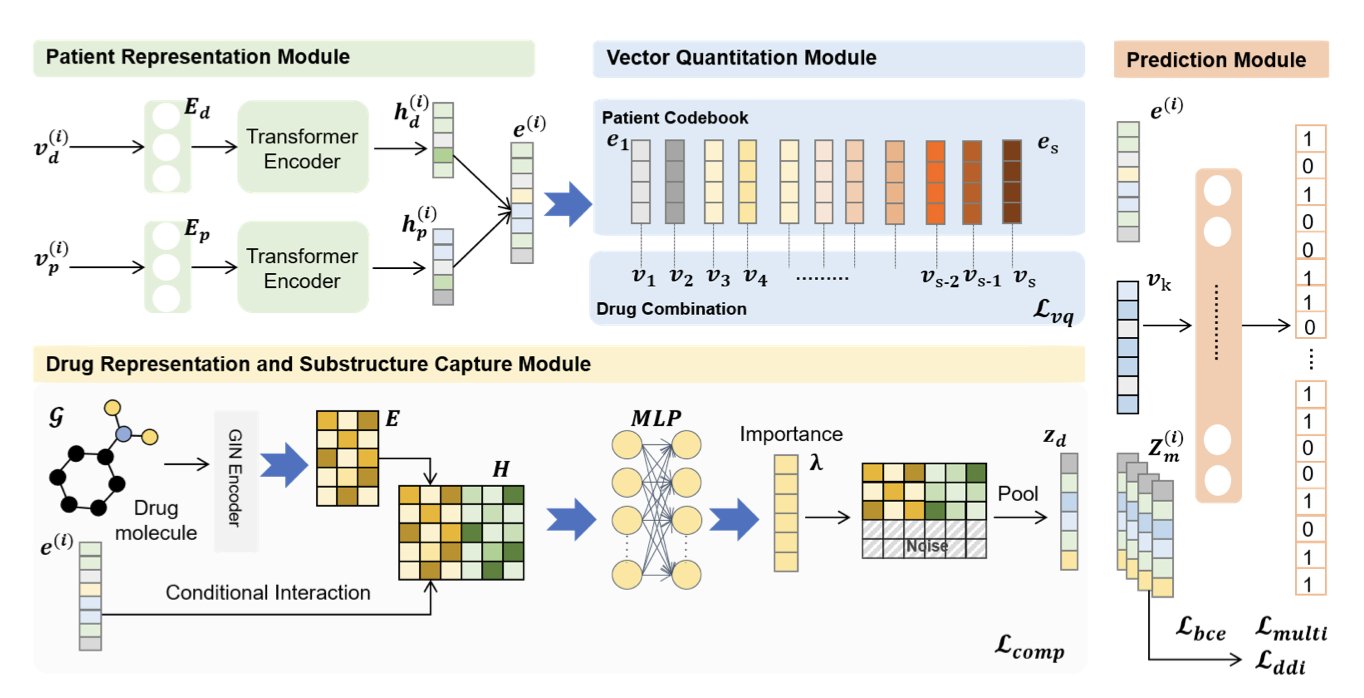
Recommending personalized medication requires balancing a patient’s complex medical history with a drug’s safety and effectiveness. Most current recommendation systems focus only on medical records, overlooking the chemical properties of the drugs themselves.
To address this, researchers developed the SubRec model. Its core idea is to connect the chemical substructures of drugs with a patient’s Electronic Health Records (EHRs). A drug’s therapeutic effects come from specific functional fragments in its molecular structure. SubRec works to find the relationships between these key fragments and specific medical conditions.
SubRec uses a conditional information bottleneck technique to filter through complex drug chemical structure information and identify the substructures most relevant to a patient’s condition. This makes the drug recommendations not just data-driven, but also helps doctors understand the mechanism of action, avoiding a “black box” recommendation process.
The model also applies an adaptive vector quantization mechanism. This technique groups vast amounts of “patient-drug” pairing data into a few representative “prototypes,” like summarizing many treatment plans into a few classic models. This reduces the computational load for model training and makes the recommendation process more controllable.
Tests on the large-scale MIMIC III and IV medical datasets showed that SubRec’s recommendations were highly consistent with doctors’ prescriptions. They also carried a lower risk of potential drug-drug interactions, improving medication safety. The model remained stable even when patient history was limited.
SubRec offers a new approach to personalized drug recommendations. Combining the chemical nature of drugs with patient clinical data is how we can achieve smart, responsible recommendations and advance precision medicine.
📜Title: Integrating Drug Substructures and Longitudinal Electronic Health Records for Personalized Drug Recommendation 🌐Paper: https://openreview.net/pdf/cfeb68d239f6c491e79abd9c1456eb7a7a3cd836.pdf
2. GeoPep: A Geometry-Aware Model for Predicting Protein-Peptide Binding Sites
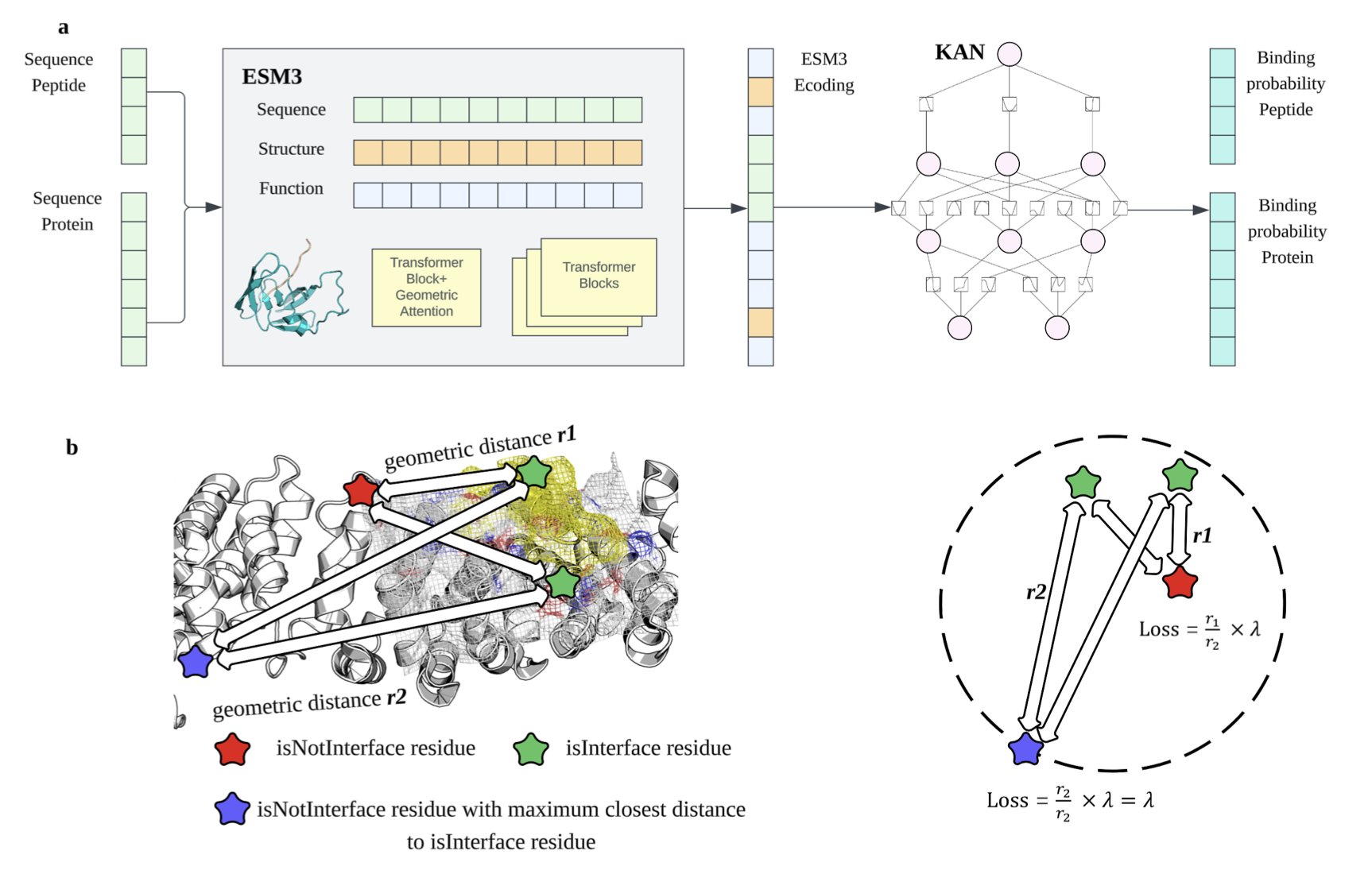
In drug development, finding peptide binding sites on proteins is difficult when high-quality structural data is limited. Predictions are challenging because often only the protein’s primary sequence is available, not its precise 3D structure. A new tool called GeoPep aims to solve this problem.
GeoPep’s main idea is to use the power of ESM3, a multimodal protein foundation model. ESM3 has learned from massive amounts of protein sequence and structure information, giving it a deep understanding of protein language. GeoPep leverages ESM3’s powerful pre-trained representations and fine-tunes them for the specific task of predicting protein-peptide binding sites.
To help the model learn complex binding patterns more efficiently from limited data, the researchers used Kolmogorov-Arnold Networks (KANs). KANs are more parameter-efficient, meaning they can capture more complex relationships with fewer parameters. In cases with sparse data, this helps prevent the model from overfitting and improves prediction accuracy.
Predicting a binding site involves more than just identifying the amino acids involved; it also requires knowing their spatial relationship. A reliable binding site is usually a continuous surface formed by amino acid residues that are close to each other in 3D space. To ensure the predicted results are geometrically reasonable, GeoPep includes a distance-based loss function during training. This function penalizes combinations of amino acids that are predicted as a binding site but are far apart in space. This ensures the final predicted site is continuous and realistic in its 3D structure.
Evaluation metrics show that GeoPep outperforms existing methods. It can accurately identify peptide-binding regions on proteins without explicit structural input, which has potential for designing new peptide drugs or inhibitors.
📜Paper: GeoPep: A geometry-aware masked language model for protein-peptide binding site prediction 🌐Paper: https://arxiv.org/abs/2510.27040v1
3. AI for Protein Design: Meet DL4Proteins, a Free Interactive Tutorial
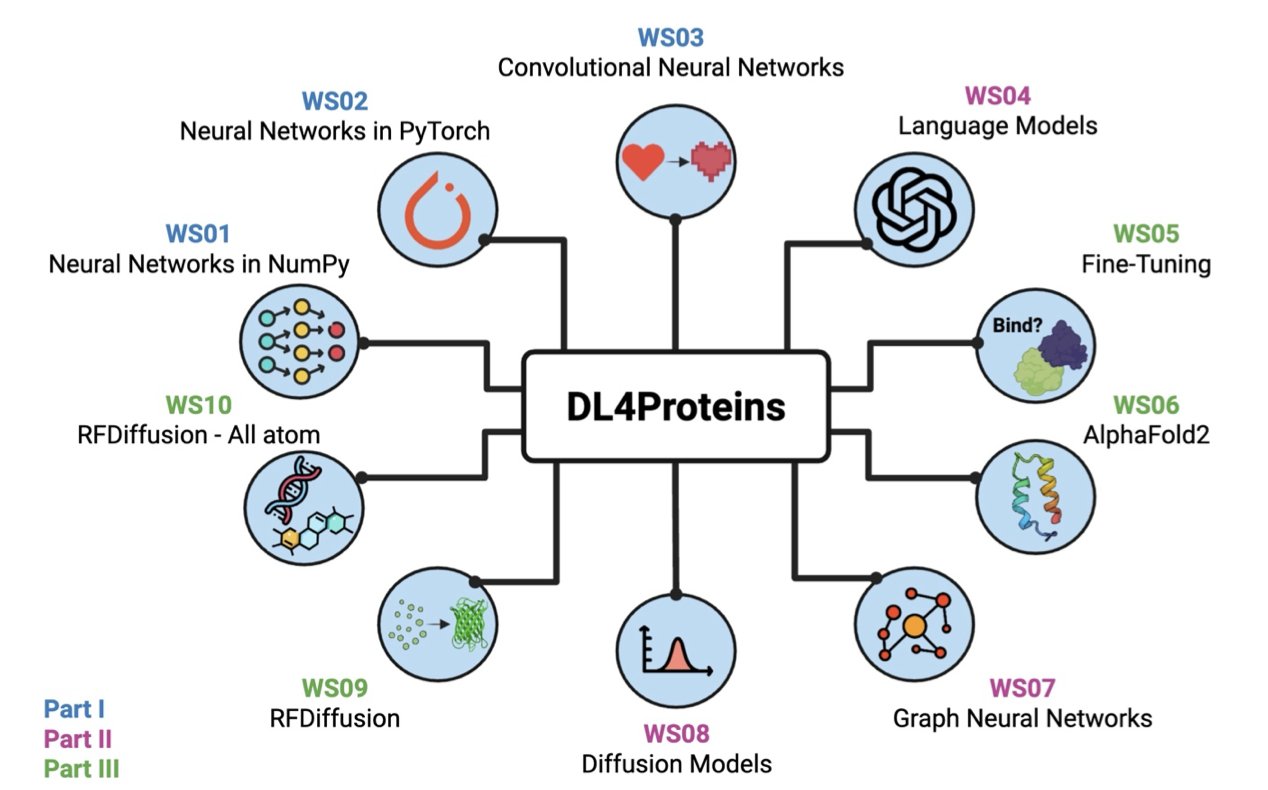
The main idea:
Artificial intelligence (AI) is becoming more important in protein structure prediction and design. But advanced AI models like AlphaFold2 often require powerful computing resources like Graphics Processing Units (GPUs), which is a barrier for many students and labs.
To solve this, researchers at Johns Hopkins University created the online tutorial DL4Proteins. They packaged complex AI models and code into interactive Jupyter Notebooks and hosted them on Google Colab. With just a browser, users can access Google Colab’s free cloud GPUs to run these advanced models. This allows them to get hands-on experience with protein structure prediction and de novo protein design.
The tutorial is structured like a well-planned course. It starts with basic concepts like neural networks to help learners understand machine learning principles. Then it moves on to deep learning architectures like AlphaFold2 and various diffusion models. This learning path works for learners from different backgrounds. Whether you are new to programming or a chemist looking to apply AI, you can get started smoothly.
Each notebook includes questions, visualizations, and practical examples to guide learners in understanding the principles behind the model’s design as they work. This hands-on approach is more effective than just reading. In a pilot course at Johns Hopkins, students with little programming experience were able to get up to speed quickly and use these tools to complete projects.
The project will be updated continuously. As new models emerge in the AI field, DL4Proteins will add new tutorials to keep the content current.
📜Paper Title: DL4Proteins Jupyter Notebooks Teach how to Use Artificial Intelligence for Biomolecular Structure Prediction and Design 🌐Paper: https://arxiv.org/abs/2511.02128v1