
Table of Contents
- Pearl achieves high-precision prediction of protein-ligand complex structures by training on massive synthetic datasets and using an architecture that understands physical symmetries. It far surpasses existing models in the sub-angstrom accuracy range that matters most to medicinal chemists.
- TriDS integrates pocket identification, conformation searching, and scoring into a single AI-native workflow, solving the inefficiency and poor physical realism of traditional molecular docking.
- MuMo fuses unstable 3D conformations with reliable 2D topological structures to provide a more stable and powerful foundation for molecular representation learning.
1. Pearl AI: Placing Every Atom with Precision, Surpassing AlphaFold 3
Protein-small molecule docking has been a tough problem in drug discovery for decades. Existing software is often based on old ideas, leading to inaccurate predictions that waste a lot of time and computing power.
The Genesis Molecular AI team developed the Pearl model to fix this.
First, Pearl addresses the problem of data scarcity. Model training usually relies on experimental data from the PDB database, which is limited and biased toward popular targets. Pearl uses a straightforward strategy: it trains on massive amounts of synthetic data. This allows the model to learn the underlying principles of physics and chemistry, not just memorize known binding patterns. In theory, this makes it better at making predictions for new targets or new chemical scaffolds.
Second is the model’s architecture. Pearl includes an SO(3) equivariant diffusion module, which has the rules of 3D space built into it. A molecule’s properties don’t change when it rotates in space. Many older models have to struggle to learn this basic fact from data, but Pearl writes “rotational invariance” directly into its code. This lets the model focus its resources on capturing the subtle interactions that determine binding, producing conformations that are more physically realistic and avoiding simple mistakes like atoms clashing.
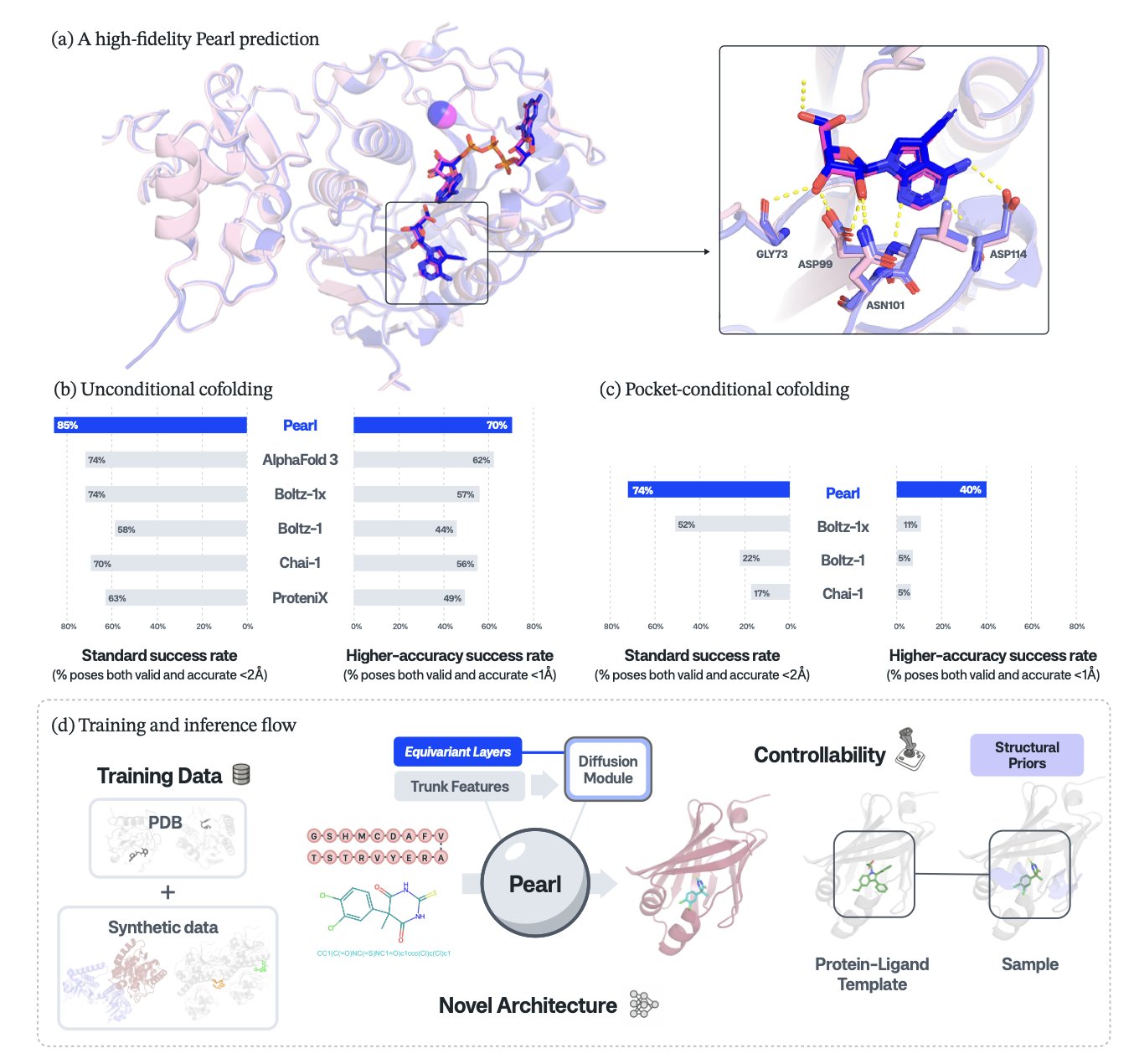
Researchers compared Pearl with top models like AlphaFold 3 on several public benchmarks. In the PoseBusters test, which emphasizes physical plausibility, Pearl achieved a success rate of 84.7%.
For medicinal chemists, the most important result is this: at the high-precision standard of RMSD less than 1Å, Pearl performs nearly 4 times better than other models.
In drug design, an accuracy of less than 1Å RMSD is critical. It can show the presence or absence of a single hydrogen bond or the orientation of a benzene ring. A 2Å RMSD conformation gives only a vague idea, but a sub-1Å conformation can tell a chemist exactly which group to optimize or which pocket to fill. This improvement in precision is a leap forward for drug design.
Finally, Pearl supports a “template” feature. If you don’t have the structure of the target protein with your ligand, but you do have its structure with a similar ligand or the structure of a related protein, Pearl can use that information as a template to improve its prediction. This feature fits well with the actual workflow of drug development.
Pearl’s design, from its data source and architecture to its applications, was carefully planned to solve fundamental problems in drug docking. How it performs on real industry projects, especially with complex and imperfect data, will be the next thing to watch.
📜Paper: A Foundation Model for Placing Every Atom in the Right Location 🌐Paper: https://arxiv.org/abs/2405.18973
2. TriDS: AI-Native Molecular Docking, Unified and Efficient
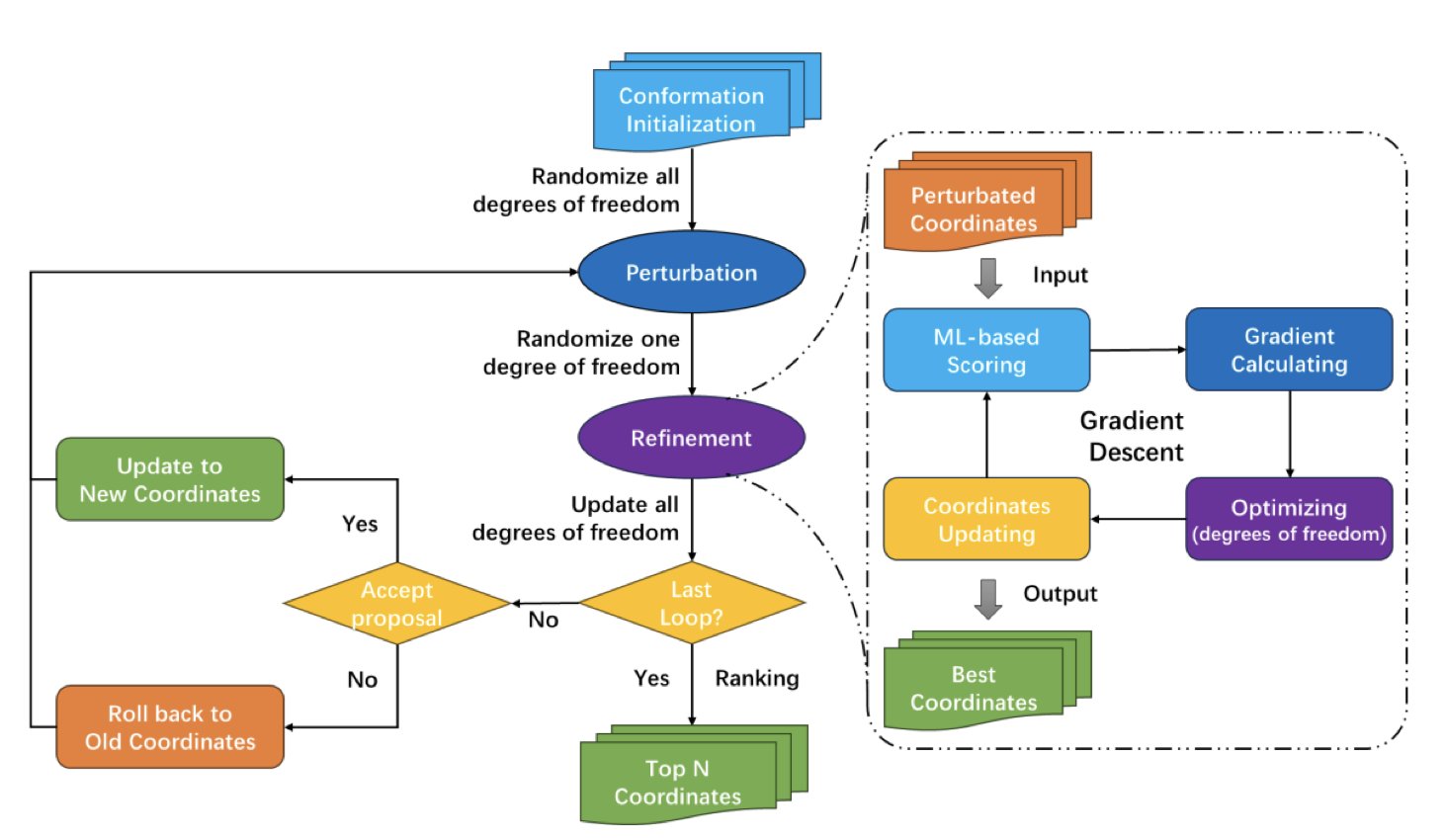
Traditional molecular docking is a multi-step process. First, you use one program to find a binding pocket on the protein. Then, you use another to try out different poses of the small molecule (conformation sampling). Finally, you might use yet another scoring function to pick the best pose. The steps are linked, and a mistake at any point can ruin the result. It’s like a multi-stage rocket where any single failure causes the mission to fail.
TriDS aims to combine this multi-stage process into a single, efficient, AI-native framework.
Here’s how it works
At the core of TriDS is a differentiable scoring function based on machine learning. It acts like a smart navigation system. Traditional docking methods are like searching in the dark, randomly trying different molecule poses and then checking how good they are. TriDS’s navigation system uses “gradient” information from the scoring function to guide the molecule to move and rotate toward lower-energy, more stable binding positions. This goal-directed search makes finding the right conformation much more efficient.
Solving a common problem in AI docking
Machine learning-only docking models sometimes generate poses that have high scores but are physically impossible, like atoms being too close together and “clashing.” This must be avoided in simulations.
The clever part of TriDS is that it adds analytic scoring function terms to its machine learning scoring function specifically to penalize atomic clashes. This is like giving the navigation system a “do not collide” command, ensuring that the generated poses are physically realistic and making the AI’s results more trustworthy.
Especially good for large molecules
Large-molecule drugs like PROTACs and macrocycles are becoming more common. Traditional docking tools struggle with them because they have so many possible conformations. Tests show that TriDS is better at docking these flexible large molecules, both in accuracy and in the physical realism of the poses it generates, compared to other non-rescoring methods. This is good news for the discovery of next-generation drugs.
From a practical standpoint, TriDS is computationally efficient, uses little GPU memory, and runs fast. It also supports various file formats and has a high success rate in parsing them. These features make it a practical tool for daily research and development, not just something for publishing papers.
📜Title: TriDS: AI-native molecular docking framework unified with binding site identification, conformational sampling and scoring 🌐Paper: https://arxiv.org/abs/2510.24186v1
3. MuMo: Fusing 2D/3D Molecular Structures to Solve a Core Problem in AI Drug Discovery
A molecule’s 3D conformation contains the spatial information that determines its function. But molecules are flexible, and a single snapshot of a low-energy conformation doesn’t tell the whole story. It’s risky for a model to train on a single, possibly unreliable, 3D structure. The MuMo framework was designed to solve this problem.
Its core idea is to combine unreliable 3D information with the reliable 2D topological structure (the molecule’s connection graph) to create a more robust “structural prior.”
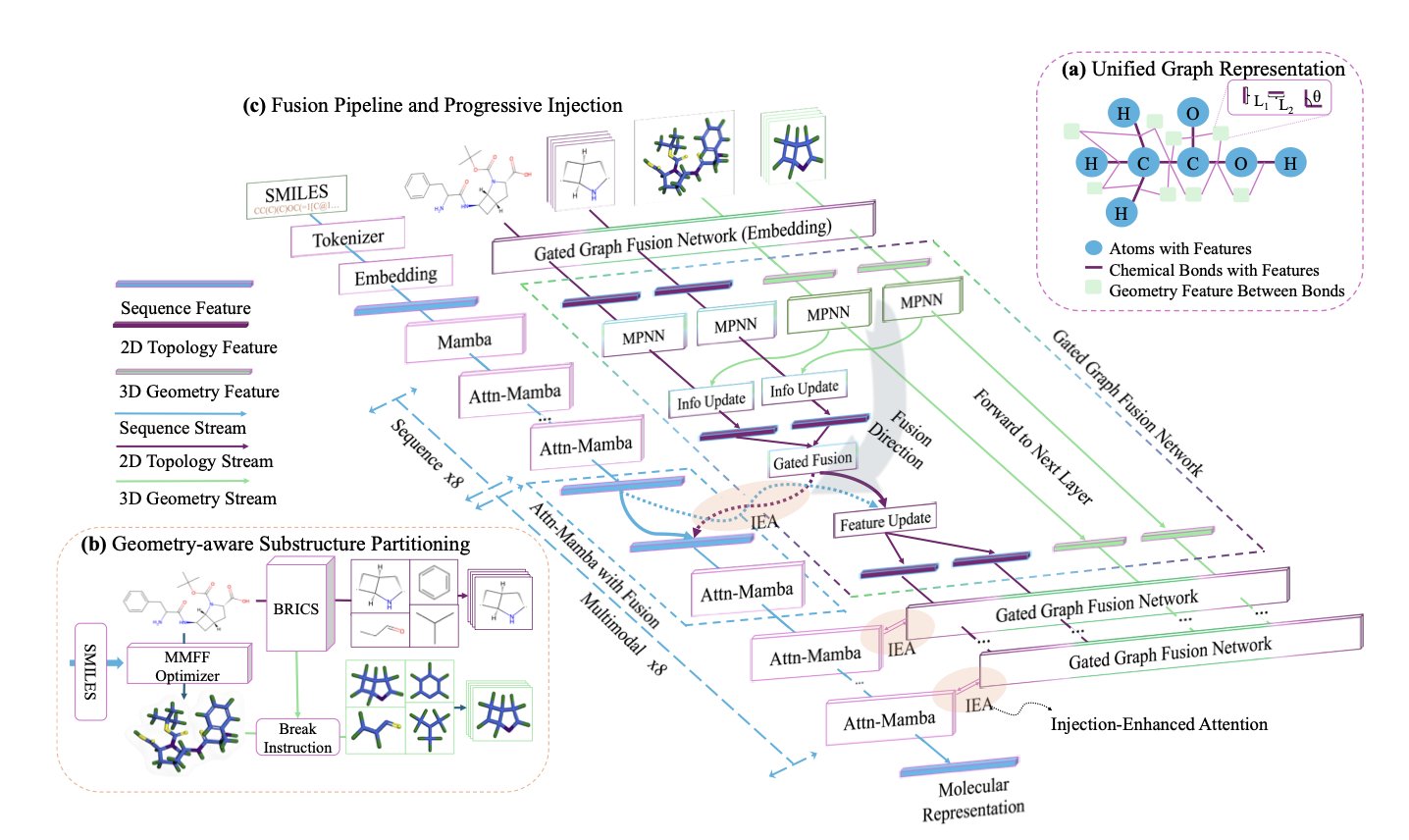
Step one: Create a more reliable molecular “blueprint”
The researchers designed a Structured Fusion Pipeline (SFP) module. It works like this: 1. It takes the molecule’s 2D connection graph and a set of 3D coordinates. 2. Through a two-step message-passing mechanism, it sends information through both the 2D graph and the 3D space at the same time. This is like having both a city road map (chemical bonds) and knowing the precise GPS coordinates of each intersection (atoms) and the distances between them. 3. The SFP merges these two types of information to create a single, stable structural representation. This representation includes both how atoms are connected and how they are arranged in space, but without relying too heavily on any one specific 3D conformation. This results in a molecular “blueprint” that is more reliable than using either 2D or 3D information alone.
Step two: Gradually inject structural information into the model
Once you have a high-quality blueprint, the question is how to use it effectively. If you feed all the structural information into a sequence model (like one that processes SMILES strings) from the very beginning, it can interfere with the model’s ability to learn from the sequence itself. This can cause “modality collapse,” where the model relies too much on one source of information.
MuMo uses a Progressive Injection (PI) mechanism. This is an asymmetric process: first, the sequence model learns for a few layers to understand the basic grammar and context of the SMILES string, like a student first learning words and sentence structures. Then, the structural blueprint generated by the SFP is injected layer by layer into the sequence model. The structural information thus acts as a higher-level guide, enriching and correcting the knowledge the sequence model has already acquired, rather than dominating the learning process from the start.
How well does it work?
On 29 benchmark tasks across TDC and MoleculeNet, MuMo improved average performance by 2.7% and ranked first on 22 of them.
The model performed best on the LD50 (median lethal dose) prediction task, with a 27% performance boost. LD50 is a classic measure of toxicity that is difficult to predict and is sensitive to a molecule’s 3D shape. The significant improvement on this task shows that MuMo’s fusion strategy gets to the heart of the matter, giving the model a deeper and more robust understanding of molecular structure.
MuMo’s design is clear. By fusing 2D and 3D information, it creates a stable representation that is not sensitive to noise in 3D conformations. This approach is a valuable reference for developing more reliable ADMET prediction models and other structure-activity relationship models.
📜Title: Structure-Aware Fusion with Progressive Injection for Multimodal Molecular Representation Learning 🌐Paper: https://arxiv.org/abs/2510.23640v1