
Table of Contents
- MetaPepticon is a fully automated bioinformatics pipeline that efficiently screens for novel Anticancer Peptides (ACPs) from massive microbial genomic data. It integrates multiple prediction algorithms to improve discovery efficiency.
- M-GLC enhances molecular property prediction in few-shot scenarios by merging global chemical motif knowledge with local neighbor molecule information.
- Interpretable machine learning models, by integrating antibody structural information, can accurately predict key properties and reveal their mechanisms of action.
1. Mining the Microbiome: MetaPepticon Automates Anticancer Peptide Discovery
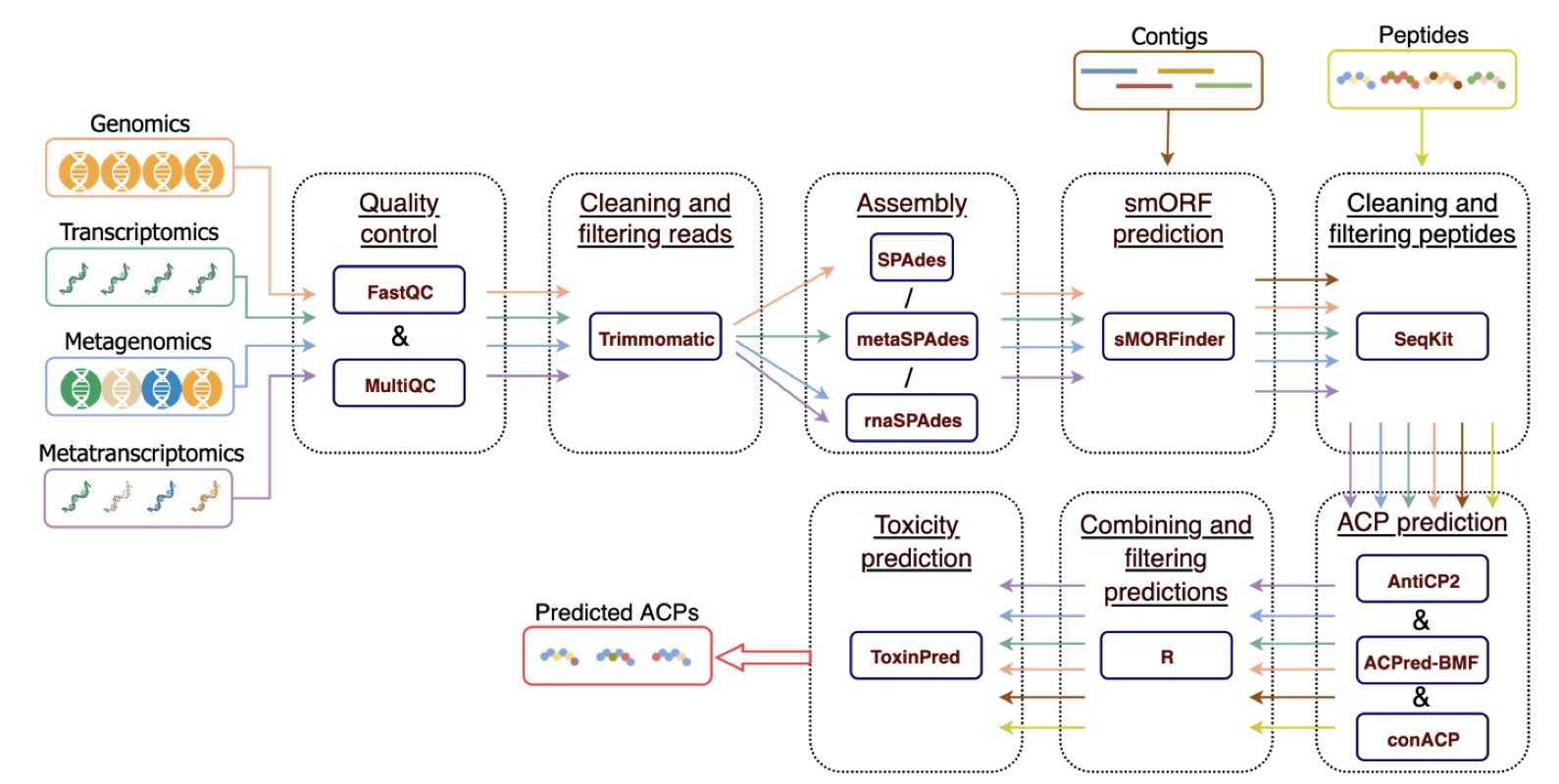
The microbiome is a treasure trove of bioactive peptides, but finding specific molecules is like searching for a needle in a haystack. Traditional bioinformatics methods require manually combining multiple tools, making the process slow, inefficient, and error-prone.
MetaPepticon streamlines the entire process of discovering anticancer peptides into a single, automated pipeline.
Here is how it works:
First, a user inputs raw sequencing data—from genomes, metagenomes, or transcriptomes. The pipeline automatically handles preprocessing, including data quality control.
The core of the process is prediction. MetaPepticon uses three independent prediction tools—AntiCP 2.0, ACPred-BMF, and ConACP—simultaneously, like a panel of experts. A peptide is only selected as a candidate if multiple tools (the threshold is adjustable) agree it has anticancer potential. This consensus strategy reduces false positives and makes the results more reliable.
Finally, the pipeline performs a toxicity prediction to automatically remove molecules that could be toxic to human cells. This ensures the clinical potential of the candidates.
The research team used the pipeline to analyze 41,171 microbial genomes and over 4 million peptide sequences. They identified 79,587 new candidate anticancer peptides. Among them, 13,149 were high-confidence candidates that passed a strict screening for known toxicity.
These results show that MetaPepticon is a scalable discovery engine. Drug developers can use it to quickly obtain high-quality lead compounds from massive microbial datasets, shortening the early discovery timeline.
The tool is open-source. Each module is packaged in a separate Conda environment to ensure the reproducibility of research results, opening up new possibilities for exploring microbial peptide libraries.
📜Title: MetaPepticon: automated prediction of anticancer peptides from microbial genomes and metagenomes 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.28.685052v1
2. M-GLC: Using Chemical Motifs to Solve Few-Shot Prediction
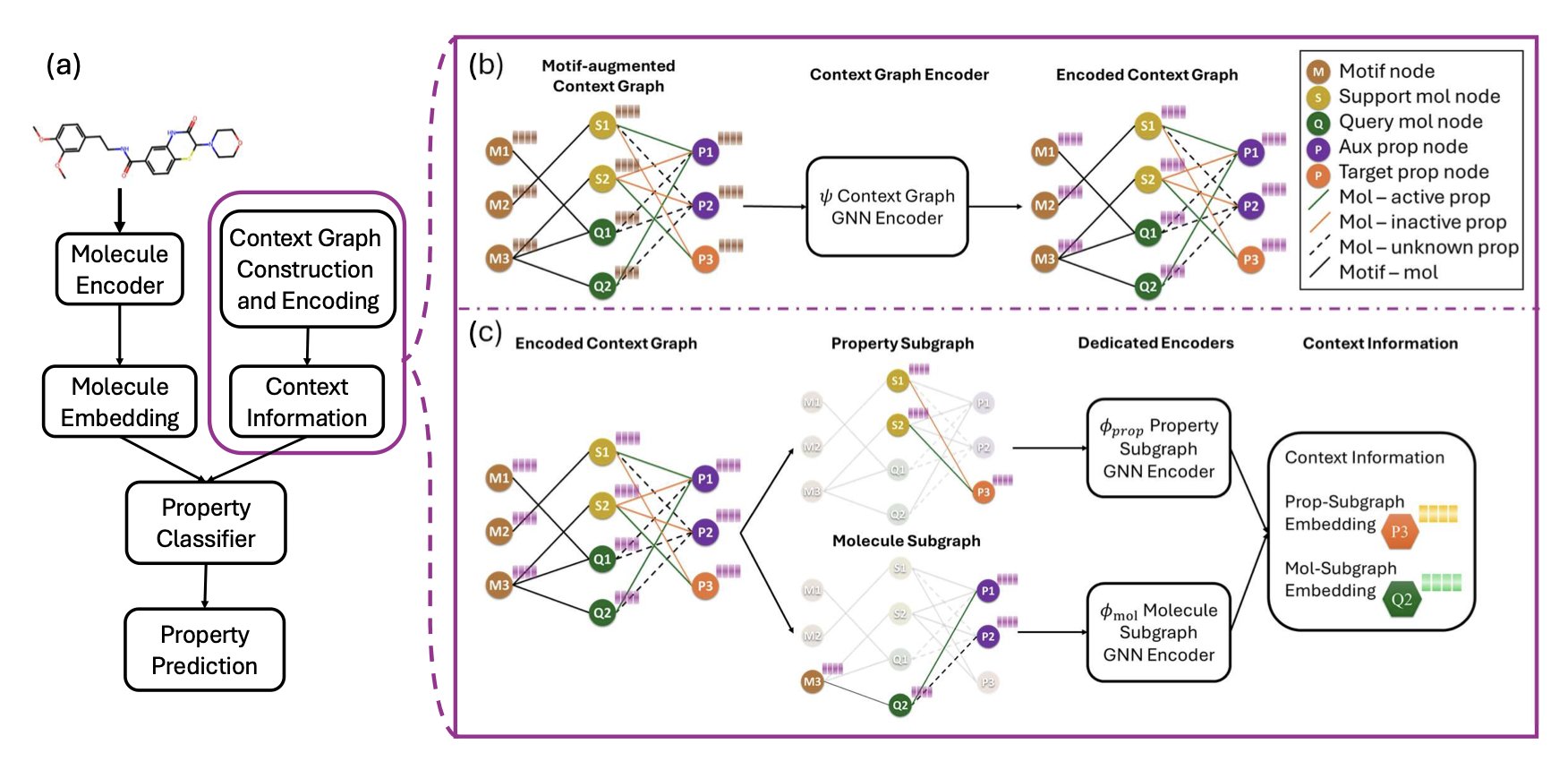
In the early stages of drug discovery, data is always scarce. Researchers might test only a few dozen compounds for activity against a new target before needing to decide on the next optimization step. This “few-shot” problem is a major challenge for computational models that predict molecular properties. Researchers at Iowa State University have proposed the M-GLC method to address this issue.
M-GLC’s core idea is to teach the model a chemist’s “chemical intuition”—the ability to recognize functional groups and scaffolds. When chemists see a new molecule, they break it down into familiar fragments (motifs) and infer the whole molecule’s properties based on the known properties of these fragments. M-GLC mimics this process.
The researchers built a tri-partite heterogeneous context graph. This graph is a network of relationships containing three types of nodes: molecules, chemical motifs, and properties. For example, a kinase inhibitor molecule would be linked to its core scaffold, a “quinazoline” motif, and also to the “high activity” property. Through this network, the model learns the connection between the “quinazoline” scaffold and “high activity.” When it encounters a new molecule containing a “quinazoline” scaffold, the model can make a prediction based on this learned association. This achieves knowledge transfer.
M-GLC combines global and local perspectives. The global perspective learns general chemical principles from the relationships between all molecules and motifs, much like reading a chemistry textbook. But the local properties of a structure-activity relationship (SAR)—where the rules for one scaffold may not apply to others—require a different view. To capture this, M-GLC introduces a local perspective by building a local-focus subgraph. This subgraph only considers the neighbor molecules most similar to the target molecule and the motifs they share. This is similar to analyzing SAR data only from a specific chemical series during an optimization campaign. The combination of global and local design allows the model to grasp general rules without ignoring local details.
Technically, the number of motif nodes in the graph far exceeds the number of molecule nodes, which creates a signal imbalance during message passing. To fix this, the model uses a structure-aware edge-weighted aggregation mechanism to balance information from different nodes, preventing motif and molecule signals from interfering with each other.
M-GLC outperformed existing state-of-the-art methods on five standard few-shot prediction benchmark datasets. In data-sparse scenarios, explicitly encoding prior knowledge, like chemical motifs, into the model architecture is an effective technical approach.
📜Title: M-GLC: Motif-Driven Global-Local Context Graphs for Few-shot Molecular Property Prediction 🌐Paper: https://arxiv.org/abs/2510.21088
3. AI for Antibody Prediction: From Black Box to Interpretable Structural Insights
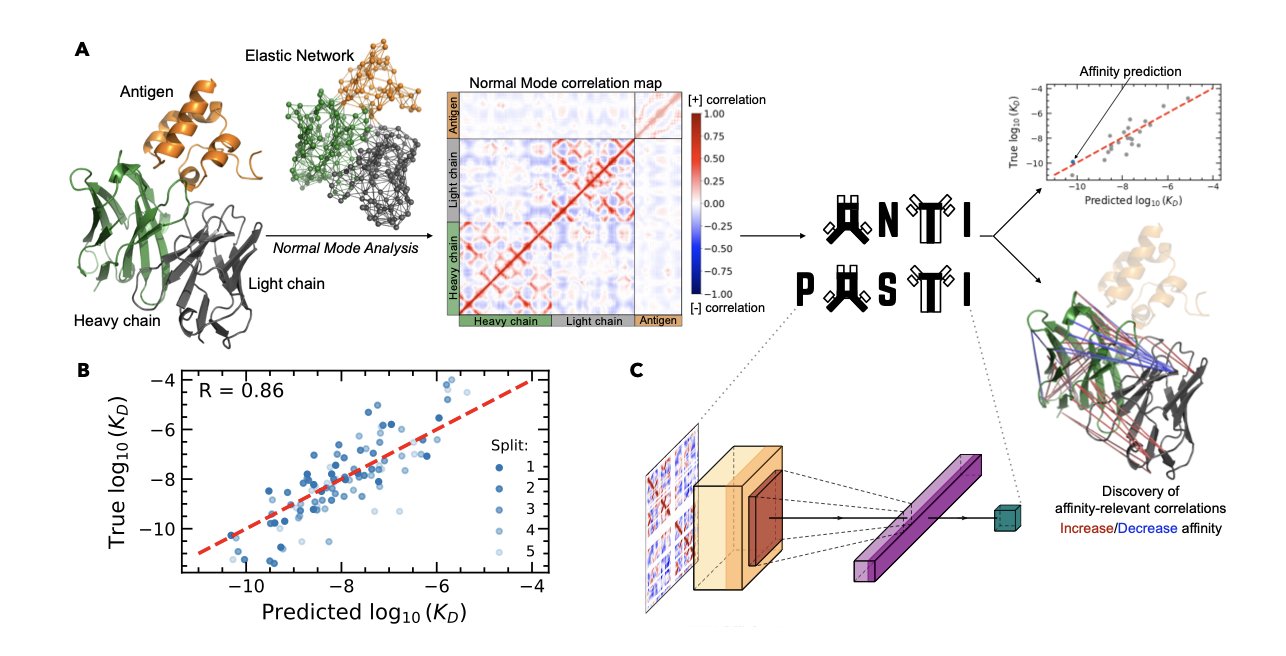
Antibody drug development requires knowing why an antibody binds. Traditional Machine Learning (ML) models are often black boxes. They give results without explaining the reasons, which is not enough for research and drug development. A new study introduces two machine learning frameworks designed to open this black box.
Researchers developed two models, ANTIPASTI and INFUSSE. They have different goals, but both integrate the 3D structural information of antibodies into the prediction process.
The ANTIPASTI model predicts the overall binding affinity between an antibody and an antigen. It treats the antibody-antigen complex as an Elastic Network Model composed of amino acids (nodes) and their interactions (springs). It then uses a Convolutional Neural Network (CNN) to learn the dynamic properties of this network, understanding the dynamic changes that occur when the complex binds. The model’s predicted affinity achieved a Pearson correlation of 0.86 with experimental data.
The INFUSSE model focuses on the flexibility of individual amino acid residues, known as the B-factor. It combines sequence information with a 3D structure-based geometric graph, processed by a Graph Convolutional Network (GCN). The model considers not only the amino acid itself but also its neighbors in space and their distances. This method predicted B-factors with a correlation coefficient of 0.71.
The key advantage of both models is their interpretability. They provide more than just a prediction value.
ANTIPASTI can perform a reverse analysis to identify the regions in the complex that contribute most to binding. The analysis points not only to the known complementarity-determining region (CDR) loops but also reveals the important role of some framework regions. This provides direct clues for protein engineering.
INFUSSE demonstrated the importance of structural information through an ablation study. When the researchers removed structural information (the graph) from the model and used only the sequence for prediction, the model’s performance dropped significantly. This shows that the behavior of an amino acid is largely determined by its 3D spatial environment.
This work advances computational methods for antibody discovery. Future tools will need to explain their prediction logic to be integrated into drug design workflows. The next step is to embed such interpretable prediction models into larger-scale automated antibody design pipelines, allowing computers to not only generate new molecules but also explain their design principles.
📜Title: Machine learning approaches for interpretable antibody property prediction using structural data 🌐Paper: https://arxiv.org/abs/2510.23975