
Contents
- The CoCoGraph model’s unique design efficiently generates chemically valid and novel molecules, a major step forward in molecular generation.
- A hybrid computational framework combines two molecular representation methods with a label-leakage-free clinical score to achieve high-accuracy, interpretable drug-drug interaction predictions.
- The DeepVul model uses transcriptome data to predict gene essentiality and drug response, providing a new tool for precision medicine that moves beyond the limitations of genetic mutations.
1. CoCoGraph: A New Benchmark for AI in Molecule Creation
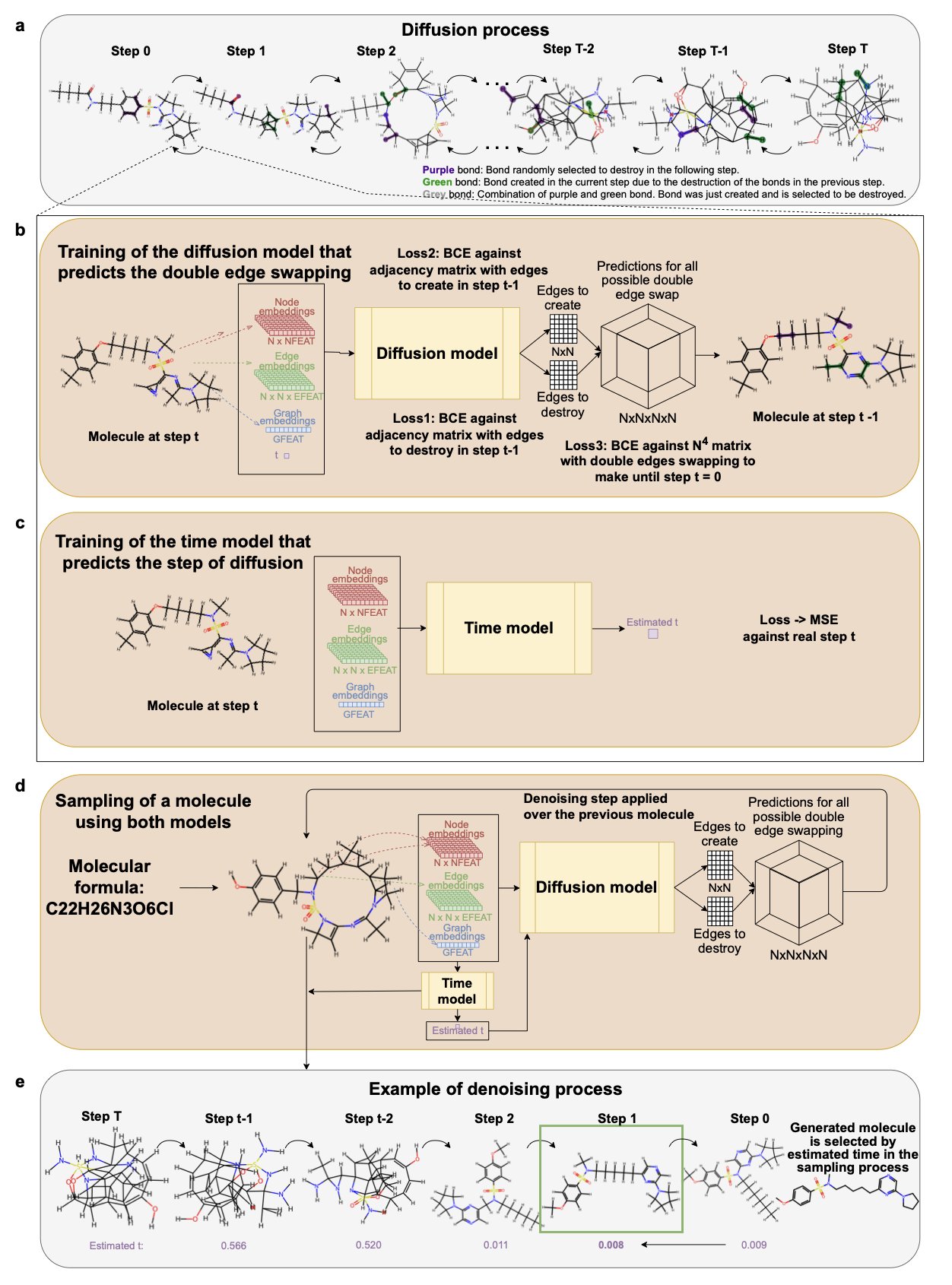
Designing new drugs or materials often requires creating entirely new molecules. In the past, generated molecules were frequently chemically invalid or not novel.
Researchers developed CoCoGraph, a collaborative graph diffusion model for generating molecules. Its design ensures that 100% of the generated molecules are chemically valid. The model uses a “bilateral exchange” method to continuously monitor atom valency, molecular formulas, and bond types throughout the generation process. These chemical rules are integrated directly into the model, so it doesn’t have to learn them on its own. CoCoGraph consists of two collaborating models: a diffusion model that predicts how chemical bonds will be exchanged, and a time model that assesses the difference between the generated graph and real molecules. Together, they improve the stability and reliability of the results.
The base version of CoCoGraph has only 534,000 parameters, an order of magnitude smaller than DiGress or JTVAE. Despite its small size, its performance is excellent. In the GuacaMol benchmark, CoCoGraph achieved 100% molecular validity, 99.9% uniqueness, and 98.6% novelty. Its Kullback-Leibler (KL) divergence for molecular property distribution was nearly half that of DiGress and over 90% lower than JTVAE, showing that its generated molecules are closer to real ones. In a comparison of 36 molecular properties, the CoCoGraph FPS version outperformed DiGress on 23 properties and JTVAE on 33. It excelled in key medicinal chemistry metrics like topology, electronic properties, and drug-likeness.
To test the authenticity of the generated molecules, researchers conducted a test where 102 experts were asked to distinguish between real molecules and those generated by CoCoGraph. The experts’ average accuracy was just 62%, slightly better than random guessing, indicating that CoCoGraph’s molecules are highly realistic and difficult to tell apart. The model can also efficiently generate millions of molecules on standard hardware. Researchers have already used CoCoGraph to build a public dataset of 8.2 million new molecules with 98.5% novelty and only 7.1% redundancy. CoCoGraph is also highly adaptable to different types of molecules. An expert evaluation found almost no difference between the distribution of acyclic or aliphatic molecules it generated and real molecules, proving the model’s ability to explore diverse chemical structure spaces.
CoCoGraph’s strength is that it integrates complex chemical rules directly into the generation process, rather than having the model learn them from data. This collaborative diffusion method with built-in chemical constraints allows for rapid, large-scale exploration of molecular space while ensuring every step follows chemical principles. It opens up a new path for molecular generation.
💻Code: http://cocograph.seeslab.net
📜Paper: https://arxiv.org/abs/2505.16365
2. A New Paradigm for DDI Prediction: Fusing Domain Knowledge with AI
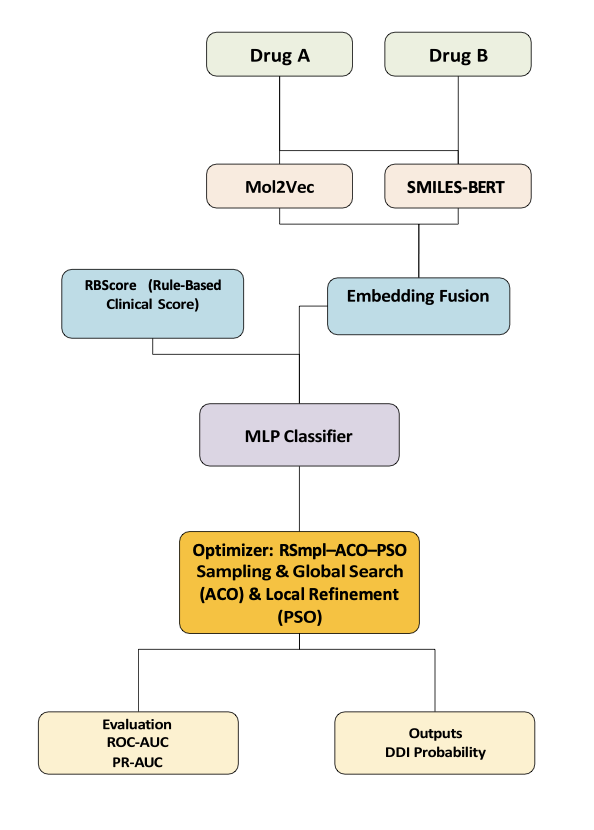
Predicting Drug-Drug Interactions (DDI) is a challenge in drug development and clinical practice. Among traditional computational methods, data-driven models act like black boxes, while rule-based systems have limited coverage. A new framework attempts to combine the advantages of both to find a balance.
First, the model understands the chemical structure of drugs through two molecular embedding techniques: Mol2Vec and SMILES-BERT.
Mol2Vec breaks down a molecule into chemical substructures, or “fragments,” and learns the relationships between them. It excels at capturing local structural information. This is like understanding a Lego model by identifying the types of bricks and how they are combined.
SMILES-BERT treats a molecule’s SMILES string as a language. It uses a Large Language Model to learn its “grammar” and context, thereby capturing the molecule’s global features. Combining the two allows the model to see both the “trees” and the “forest,” giving it a more complete understanding of drug molecules.
A core part of the framework is a clinical scoring mechanism called RBScore. A major pitfall in DDI prediction is “label leakage,” where the model is exposed to information during training that it shouldn’t have. This leads to inflated performance in tests but failure in real-world applications.
To avoid this problem, RBScore is built entirely on pharmacological knowledge, without relying on any known DDI labels. The score is calculated based on a drug’s pharmacokinetic (PK) and pharmacodynamic (PD) properties, such as whether a drug is an inhibitor or substrate of a specific CYP enzyme. This is like giving the model the built-in experience of a pharmacologist. Before making a prediction, the model consults this unbiased “expert opinion,” which improves its ability to generalize. This also enhances interpretability: when the model predicts an interaction, you can trace it back to RBScore to see which pharmacological rule was at play.
For the classifier and optimization, the framework chose a lightweight classifier instead of a complex neural network. This reduces computational cost and makes it easier to deploy in practice.
To efficiently find the best hyperparameters, the framework uses a three-stage metaheuristic optimization strategy (RSmpl–ACO–PSO). In the first stage, random sampling (RSmpl) is used to perform an initial screening of the parameter space to identify promising regions. In the second stage, Ant Colony Optimization (ACO) is used to finely explore these regions. In the third stage, Particle Swarm Optimization (PSO) performs a final local search around the best points. This strategy of moving from global exploration to local optimization balances efficiency and stability.
The model achieved a ROC-AUC of 0.911 on the DrugBank dataset and performed well on a real-world cohort of type 2 diabetes patients, validating its potential for clinical application.
This framework skillfully combines machine learning with domain knowledge to address key pain points in DDI prediction. Its modular design also leaves room for future updates, such as incorporating more advanced molecular representation methods or integrating electronic health record data to enable personalized DDI predictions.
📜Title: A Hybrid Computational Intelligence Framework with Metaheuristic Optimization for Drug–Drug Interaction Prediction 🌐Paper: https://arxiv.org/abs/2510.09668
3. DeepVul: Predicting Drug Response with a Transformer, a New Path for Precision Medicine
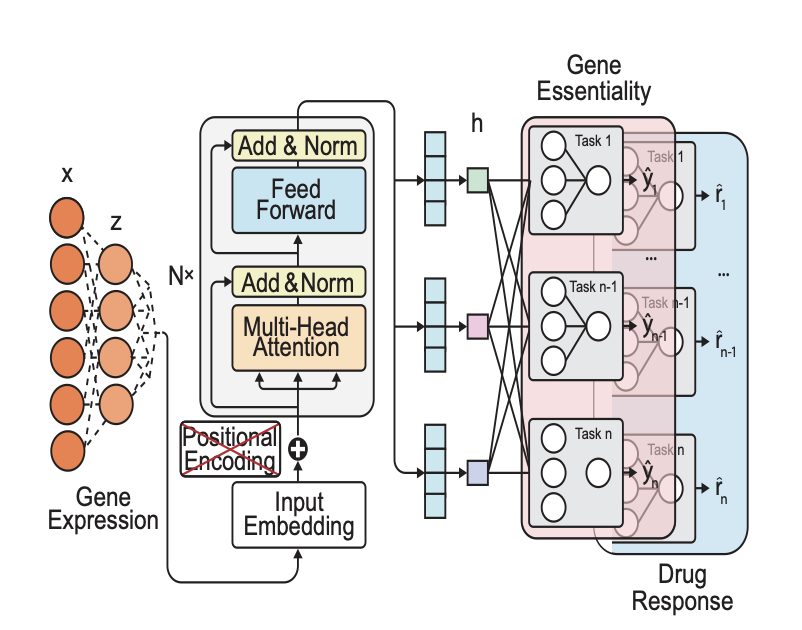
Current precision medicine mainly relies on finding specific targetable gene mutations, but this is effective for only a small number of cancer patients with such mutations. DeepVul aims to break through this limitation and find treatment options for a broader group of patients.
DeepVul’s approach is to analyze a cell’s “real-time work log”—the transcriptome, which is the activity level of all genes in a cell at a specific moment. The authors believe this log reveals a cell’s weaknesses better than static genetic mutations.
Here’s how it works
The model uses a multi-task Transformer architecture, which can be thought of as an intelligent system that learns three types of information at the same time: 1. Gene expression profiles: The activity levels of thousands of genes in a cancer cell line. 2. Gene perturbations: The changes that occur in a cell when a specific gene is “knocked out” (for example, using CRISPR technology). 3. Drug perturbations: The change in a cell’s state after it is exposed to a certain drug.
DeepVul’s core design maps these three types of information into a unified mathematical space, known as the “latent space.” This forces the model to find the common underlying biological logic among them. For instance, if the gene expression profile of cell line A is similar to that of cell line B after gene X has been knocked out, the model will infer that gene X may be cell line A’s “Achilles’ heel.”
An unexpected discovery
To test the model’s ability to generalize, the researchers conducted a transfer learning experiment. They first trained the model on the vast gene essentiality data from the DepMap project, allowing it to learn the basic survival logic of cancer cells. Then, they gave the model a new task: to predict how cells would respond to different drugs.
The test included two strategies: one was to fine-tune the entire model to adapt to the new task; the other was to “freeze” the encoder responsible for understanding gene expression profiles and only train the final part responsible for prediction.
Unexpectedly, the “freezing” strategy worked better.
This result suggests that the patterns learned from gene essentiality data are more fundamental and universal than those learned from drug response data. Gene essentiality reflects the core systems that a cell depends on for survival; it is a purer and more global signal. Drug response data, on the other hand, might contain “noise” like off-target effects. This is like learning a language: mastering the underlying grammar (gene essentiality) is more effective than just memorizing a few everyday phrases (drug response).
The model’s value
DeepVul can find potential treatment targets for more patients. Tests have shown that its accuracy in predicting sensitivity to hundreds of gene targets is comparable to existing mutation-based methods, thereby expanding the potential pool of beneficiaries.
The model is interpretable. Researchers can use tools like SHAP to investigate the basis for its decisions. For example, when predicting a cell’s sensitivity to a BRAF inhibitor, the model can identify which genes’ expression levels influenced the prediction. This is valuable for understanding drug mechanisms and resistance.
The model’s generalization ability has also been verified. A model trained on data from the Broad Institute performed robustly on data from the Sanger Institute without any retraining. This proves that the model learned universal biological principles, not just patterns specific to a particular dataset.
📜Paper: DeepVul: A Multi-Task Transformer Model for Joint Prediction of Gene Essentiality and Drug Response 🌐Preprint: https://www.biorxiv.org/content/10.1101/2024.10.17.618944v2 💻Code: https://github.com/alaaj27/DeepVul.git