
Table of Contents
- Researchers combine predictive and generative AI to design small molecules that precisely target specific cancer cells, opening a new path for phenotypic drug discovery.
- The MATCHA model uses multi-stage flow matching to balance speed, accuracy, and physical validity in molecular docking, showing practical value for drug discovery.
- A new tool uses AI to automatically link amino acid residues mentioned in papers to their 3D structures, along with local structure quality assessment data.
1. AI Designs New Anti-Cancer Drugs to Precisely Target Specific Cancer Cells
There are usually two ways to think about drug discovery. One is target-based, like using a specific key for a specific lock. The other is phenotypic screening, where you test a huge number of compounds to see which ones “kill cancer cells,” without worrying about the lock. Phenotypic screening can uncover entirely new mechanisms, but optimizing the hits is like fumbling in a black box.
This new study installs a smart navigation system inside that black box.
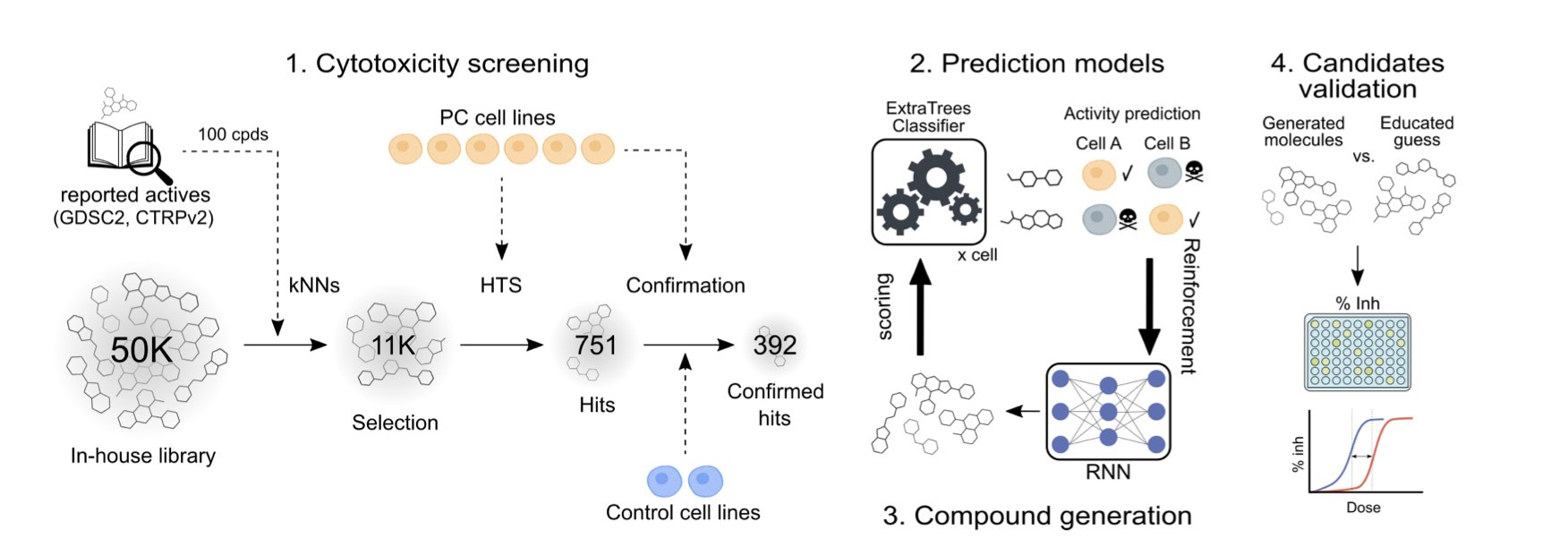
Its approach has three steps.
First is data collection. The research team treated six pancreatic cancer cell lines and two normal cell lines with over 11,000 compounds. This generated a massive amount of raw data, revealing how different compounds affect different cells.
Second is training a machine learning model to recognize the unique patterns—the “bioactivity fingerprints”—produced when a compound acts on different cell lines. The model’s goal is to find molecules that “only kill cancer cell A, but are harmless to cancer cell B and normal cells.” The results showed that this bioactivity-based model was more accurate than traditional models that only analyze chemical structures. This proves the AI understood the deeper rules of biological effects.
Third is letting the AI create new molecules. The researchers embedded their trained model into the generative AI platform REINVENT. REINVENT acts like a chemist, constantly generating new molecular structures. The discriminator model then immediately judges their selectivity, pointing out if a molecule might harm normal cells or confirming its potential and guiding the design. Through this reinforcement learning loop, the AI shifts from random screening to targeted, intelligent design. It can even design molecules with differential killing effects for cancer cell lines with similar molecular backgrounds.
The AI-designed molecules were validated experimentally. The team synthesized and tested 45 new molecules, and 20 of them showed the expected cell-specific killing effect. A hit rate of nearly 45% is far higher than the less than 1% rate of traditional high-throughput screening, proving the effectiveness of the AI’s design.
This work combines the unbiased advantage of phenotypic screening with the design power of generative AI, demonstrating a new paradigm for drug discovery. In the future, we might be able to directly design drugs that reverse complex disease phenotypes, like fibrosis or neurodegeneration, without needing to identify a target first. For tough diseases like pancreatic cancer, this approach offers a promising new strategy.
📜Title: Phenotypic AI-based design of cell-specific small molecule cytotoxics 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.15.682546v1
2. MATCHA: A Breakthrough in AI Molecular Docking That’s Both Fast and Accurate
Molecular docking has long faced a trade-off between speed and accuracy. Traditional methods are fast but not very accurate. Co-folding models like AlphaFold 3 are precise but take too long to compute, making them unsuitable for large-scale virtual screening. The new MATCHA model aims to solve this problem.
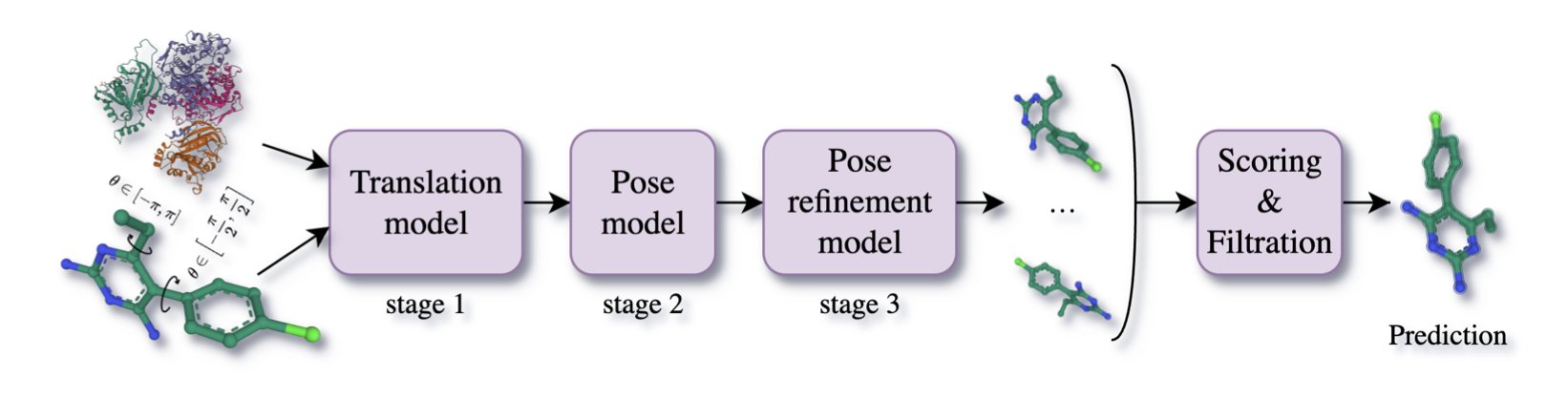
Its core is a “coarse-to-fine” multi-stage process:
- First, global positioning. The model finds a general landing spot for the ligand molecule in 3D space (R³), aiming to quickly place it near the target protein.
- Second, conformer adjustment. Once the rough position is set, the model fine-tunes the molecule’s overall orientation in SO(3) space, like aligning a key with a keyhole.
- Third, fine-tuning rotatable bonds. Finally, the model carefully adjusts the freely rotatable chemical bonds within the molecule in SO(2) space, ensuring the molecular conformation fits the pocket perfectly.
This divide-and-conquer strategy is effective. It breaks the complex docking problem down into several simpler sub-problems.
On the ASTEX test set, MATCHA achieved a success rate (RMSD ≤2Å) of 66%. The model also emphasizes the physical validity (PB-valid) of its results. Many docking programs generate conformations with low RMSD values but with unreasonable atomic distances or spatial clashes. MATCHA has a built-in unsupervised physical validity filter to screen out such results. This is valuable for drug development projects, as it can save a lot of time on manual screening.
In terms of speed, MATCHA is about 25 times faster than large co-folding models like AlphaFold 3. This improvement makes it usable for screening much larger compound libraries. The model is also cheaper to train, requiring only a single H100 GPU for 38 days, making it easier for different labs to reproduce and improve upon.
MATCHA’s model architecture combines the strengths of a Diffusion Transformer and a Uni-Mol-like spatial encoder. It can handle both blind docking and docking with a known pocket, making it widely applicable. The model still has room for improvement, such as addressing the long-standing challenge of receptor flexibility. By improving computational efficiency while maintaining high accuracy and generating more chemically reasonable results, MATCHA is a practical tool with the potential to be used on the front lines of drug discovery.
📜Title: MATCHA: Multi-Stage Riemannian Flow Matching for Accurate and Physically Valid Molecular Docking 🌐Paper: https://arxiv.org/abs/2510.14586
3. AI Reads Papers, Linking Text to 3D Protein Structures in One Click
We’ve all been there.
You’re reading an article about a new drug target, and the author mentions a key interaction, like a compound binding to “Arginine 254” on a protein. What do you do next? Open PyMOL, download the PDB file, find the right chain, and then find that residue number. Ten minutes later, your train of thought is gone.
A team has developed a tool to solve this problem.
They trained a Transformer model, a type of Large Language Model (LLM), specifically to read biochemistry literature. This AI’s job is to recognize expressions like “Arg254” or “the A254K mutation” in a sentence.
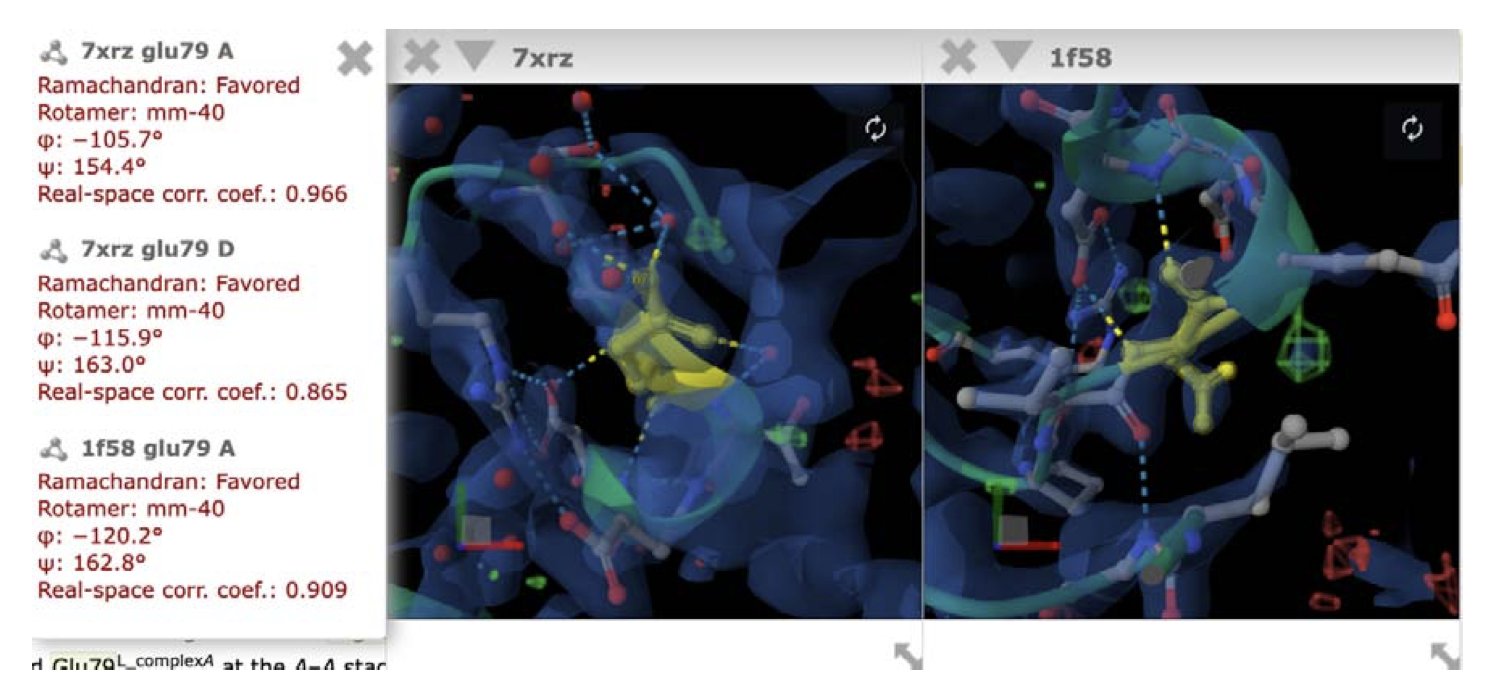
Recognition is just the first step. The tool automatically maps this text information to a sequence in the UniProt database and then links it to its 3D coordinates in the Protein Data Bank (PDB). You just click a link to see that key arginine residue in a 3D view.
This tool has another great feature: when it displays the structure, it also provides quality metrics for the local experimental data of that residue.
Anyone who does structural biology knows that the reliability of a protein crystal structure model can vary wildly from one region to another. A flexible loop area, for instance, might have a blurry electron density map. This tool will tell you directly: “The Arg254 you’re looking at has high-quality data,” or “Be careful, the model for this region is speculative and not very reliable.” This provides a direct basis for assessing the reliability of a paper’s conclusions.
It’s like a navigation app that not only shows you the destination but also warns you about road conditions and risks along the way.
You can imagine if this tool were integrated into a journal’s peer review system, reviewers could instantly verify an author’s structural analysis. For drug design, it would shorten the time needed for target validation and structural analysis. The research team also plans to expand its capabilities to identify protein names, functional states, and other information, which would make the system even more complete.
📜Paper: Linking Protein Residues in Literature and Structure 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.17.683004v1