
Table of Contents
- The M3ST-DTI model uses a two-step “coarse-to-fine” fusion strategy, integrating textual, structural, and functional data to improve drug-target interaction (DTI) prediction accuracy.
- This study uses an XGBoost model to analyze gene expression data, predicting breast cancer patients’ response to neoadjuvant therapy and matching each patient with the most effective drug.
- KG-Bench provides a standardized evaluation framework for graph neural network algorithms in drug repurposing, solving challenges of data leakage and unfair model comparisons.
1. The M3ST-DTI Model: Multimodal, Step-wise Fusion Improves Drug-Target Prediction
Predicting whether a drug molecule will bind to a target protein is central to drug discovery. Past computational models tried to speed this up, but with limited success. They often relied on a single type of information, like molecular structure or protein sequence. That approach is like trying to identify a person from just one photo—it doesn’t give you the full picture.
The M3ST-DTI model takes a different approach. It works on the idea that to accurately predict drug-target interactions, you have to intelligently use all available information.
Multimodality: Three Experts in One Room
The model describes drugs and targets from three different perspectives at once:
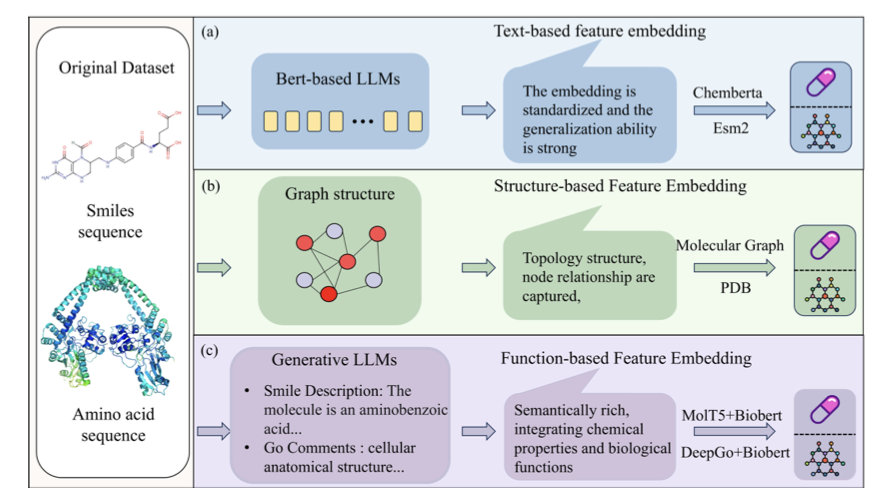
- Textual features: It treats a drug’s SMILES string and a protein’s amino acid sequence as a special kind of “language.” The model uses self-attention mechanisms to understand these sequences and capture key information, like a drug’s functional groups or a protein’s critical domains.
- Structural features: This focuses on the topology of the molecule and protein—how the atoms are connected. The model uses a graph attention module, viewing the molecule as a network of atoms and chemical bonds to extract spatial relationships.
- Functional features: This describes a protein’s role in the cell and the biological pathways it participates in. This adds high-level biological context to the interaction.
Combining these three sources of information is like getting a chemist, a structural biologist, and a cell biologist to consult on a case. The result is more complete than what any single perspective could offer.
A Two-Step Fusion: First Align, Then Refine
The biggest challenge is how to effectively merge three types of data that have different formats and scales.
M3ST-DTI uses a two-stage fusion strategy:
Step 1: Early Fusion—Getting on the Same Page In this step, the model’s goal is to “align” the feature vectors from the textual, structural, and functional modes in mathematical space. It uses Gram loss as a constraint, which acts like a coordinator, forcing the internal relationships of different feature vectors to be consistent. For example, two structurally similar drugs should also have similar textual and functional features. After this step, the three feature types are placed in a common context, setting the stage for a more precise fusion later.
Step 2: Late Fusion—Fine-Tuning Once the features are aligned, the model begins the fine-tuning process.
How Well Does It Work?
A UMAP visualization shows the model’s effect clearly. With the raw input features (left side of the image), the data points for binding (positive samples) and non-binding (negative samples) are mixed together and hard to separate. As the model progressively fuses and optimizes the data, these points gradually separate, eventually forming two distinct clusters (right side of the image). This shows the model learned to effectively tell the two cases apart.
On the public BindingDB dataset, M3ST-DTI outperformed current state-of-the-art methods on metrics like accuracy, F1 score, AUROC, and AUPRC. This demonstrates that the “multimodal input + step-wise fusion” architecture is effective. It solves a key problem in DTI prediction through a better feature integration process.
📜Title: M 3ST-DTI: A Multi-Task Learning Model for Drug-Target Interactions Based on Multi-Modal Features and Multi-Stage Alignment 🌐Paper: https://arxiv.org/abs/2510.12445
2. AI Accurately Predicts Breast Cancer Treatment Response, Optimizing Personalized Therapy
In cancer treatment, a key goal is to match each patient with the most effective drug. For breast cancer patients receiving neoadjuvant therapy (NAT), if the tumor disappears completely before surgery—achieving a pathological complete response (pCR)—their chances of long-term survival improve greatly. But right now, drug selection often relies on trial and error.
Researchers used the XGBoost machine learning algorithm to address this problem. XGBoost is good at identifying patterns in complex data.
Here’s how the model works:
First, the researchers used high-quality data from the I-SPY2 clinical trial. This is a large, well-designed study that includes data on various new drug regimens and the corresponding gene expression of patients. The quality of the data sets the upper limit for the model’s performance.
Then, the model began to learn. It analyzed the expression levels of thousands of genes, looking for connections between high or low gene expression and whether a patient achieved pCR. It can examine the complex relationships between hundreds or thousands of genes at the same time.
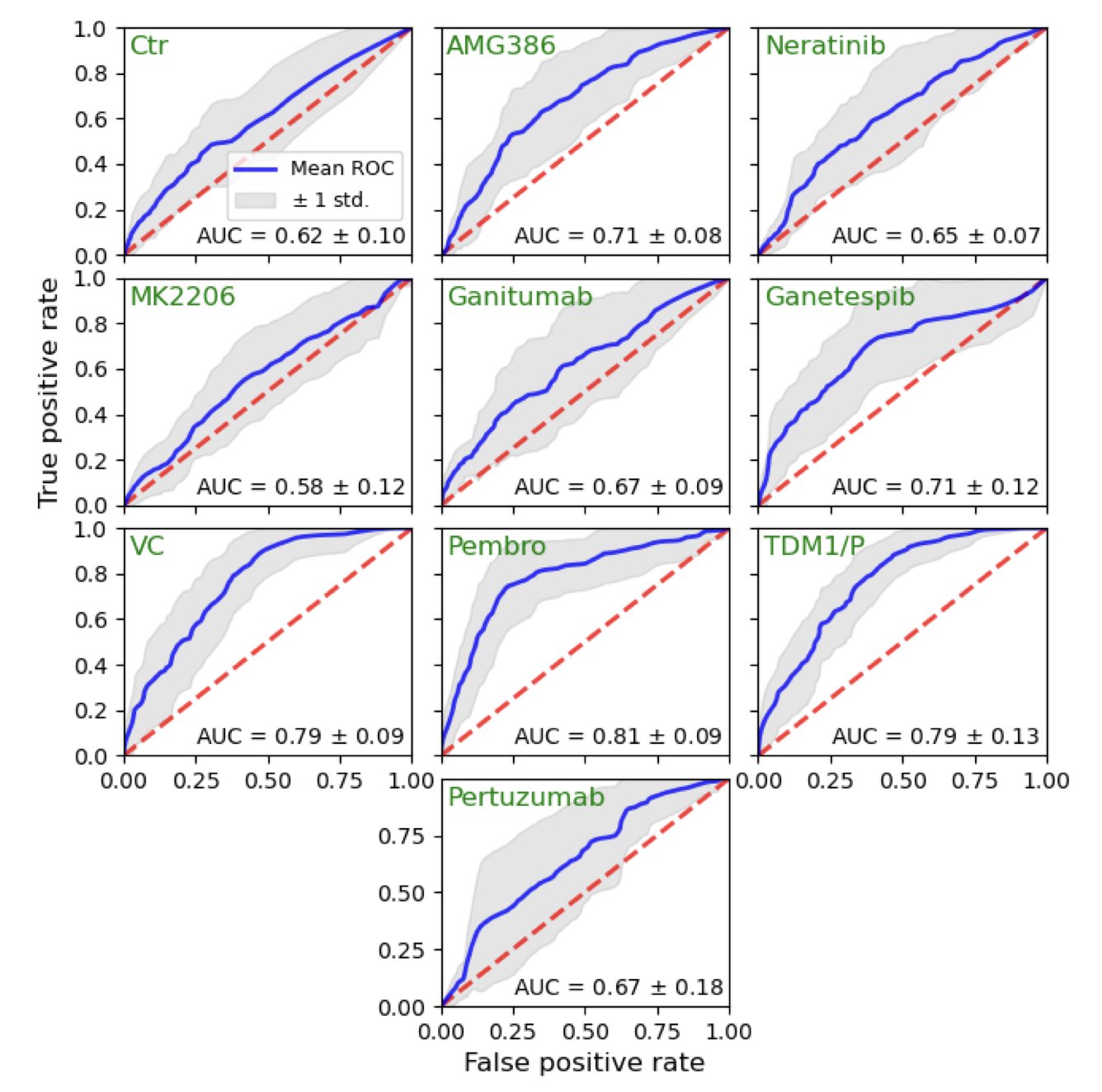
The results showed the model performed well. For some treatment regimens, its predictive accuracy (measured by the AUC metric) reached 0.814. In biomedicine, an AUC above 0.8 is considered a reliable prediction. This means the model can effectively distinguish which patients are likely to respond well to a specific drug.
The model also found some biological clues. For example, patients with low expression of the ABDH1 gene or high expression of the DENND1C gene seemed more likely to benefit from treatment. These findings offer new ideas for drug development, could help in studying drug resistance mechanisms, and might even lead to the discovery of new drug targets.
Finally, this work shows the model’s potential for clinical use. The researchers set a probability threshold—for example, only giving a specific drug to patients with a predicted pCR probability over 50%. Simulations showed that with this strategy, the pCR rates for drugs like TDM1/P and Pembrolizumab could be increased. This is a form of precise drug guidance, getting the right drug to the patients who need it most.
Of course, the study is limited by its sample size. All machine learning models need to be validated in larger, more diverse populations to confirm they are robust. But it points the way forward: using AI to interpret a tumor’s genetic information can advance personalized cancer therapy.
📜Title: Machine Learning for Predicting and Maximizing the Response of Breast Cancer Patients to Neoadjuvant Therapy 🌐Paper: https://doi.org/10.1101/2025.10.11.25337587
3. KG-Bench: A Fair Competition for GNN Models in Drug Repurposing
In AI drug discovery, one persistent problem is figuring out which model is actually better. Every team claims its algorithm is the best, but they use different datasets and evaluation methods. It’s like trying to compare people’s height using different rulers on different playing fields—the comparisons aren’t fair.
KG-Bench was built to solve this. It is a public “arena” that provides a fair, standard environment for graph neural network (GNN) models used in drug repurposing.
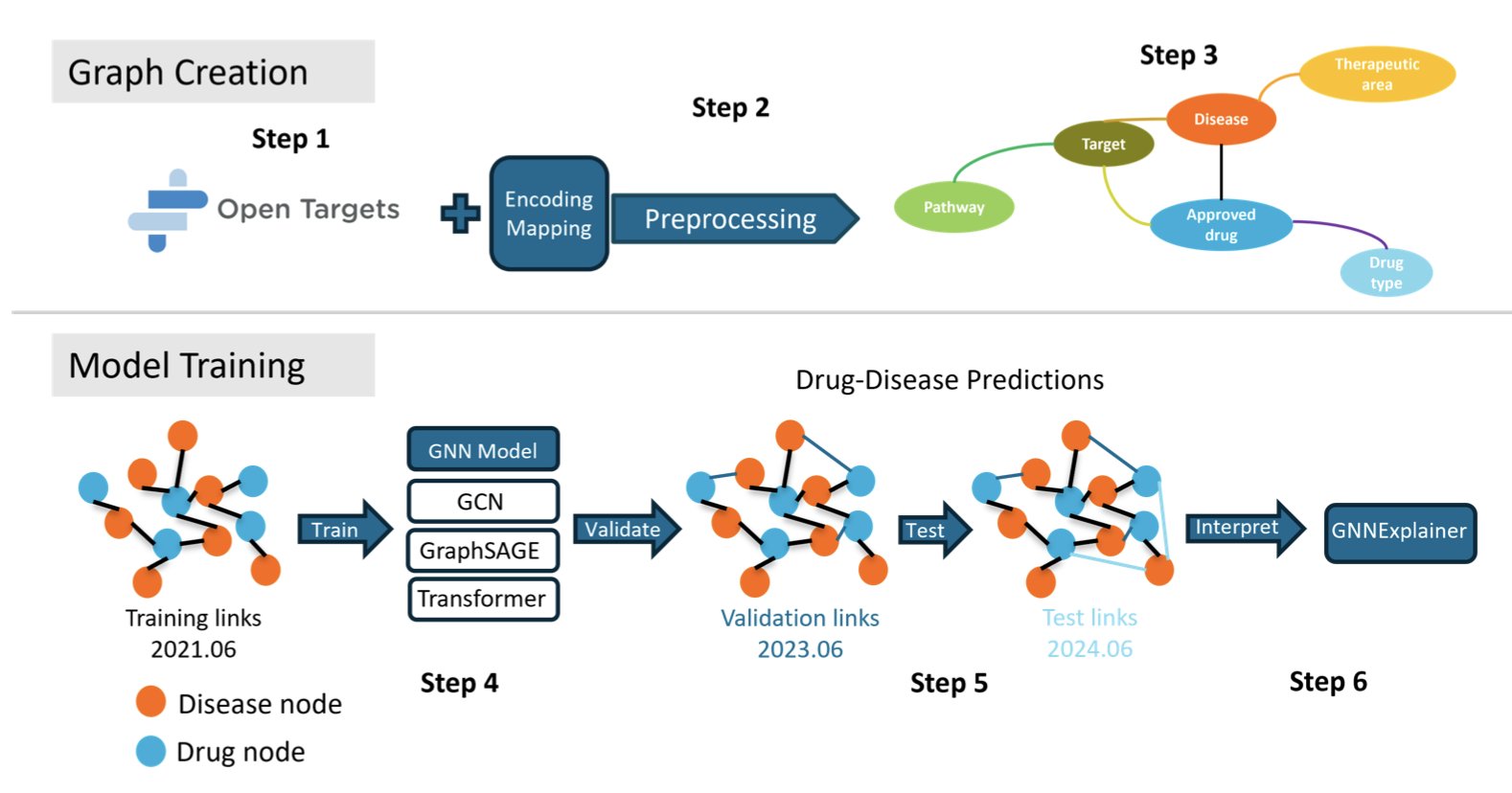
A fair competition starts with a standard playing field. KG-Bench chose the industry-recognized Open Targets dataset and used it to build a massive knowledge graph (KG) with information on drugs, diseases, targets, and biological pathways. This graph serves as the standard “exam” for all models, ensuring the data source is consistent.
Next, it tackles a key problem: data leakage, which is a form of “cheating.” In drug repurposing, a common pitfall is using future knowledge to predict the past, which can inflate a model’s performance. KG-Bench prevents this with retrospective validation, which involves a strict chronological split of the data. This ensures that when a model makes a prediction, it hasn’t seen any information from “after” the event. This way, all models start from the same line, and their results are trustworthy.
In this fair arena, several GNN architectures were tested. The results showed that the TransformerConv model performed best, with an average precision (APR) of 0.87. The core of a Transformer is its attention mechanism, which is good at capturing key information in complex relational networks. Since biological systems are complex networks, this helps explain its strong performance.
The value of KG-Bench goes beyond just performance scores.
It also analyzes the models for potential biases. The study found that “star” drugs or diseases—those with more connections in the training data—were more likely to get high-ranking recommendations from the models. This suggests that models may be biased toward recommending well-researched “familiar faces” while overlooking less-studied but potentially valuable options. In real-world applications, this kind of bias needs to be identified and corrected.
The framework also uses the GNNExplainer tool to open up the algorithm’s “black box.” When a model predicts that a drug might treat a certain disease, the tool can show which key targets or pathways the model based its decision on. This gives biological logic to a prediction score, which gives researchers more confidence in using the results.
KG-Bench is doing the work of “paving the road.” It provides an open-source, modular framework where anyone can bring their new model and test it against the current best ones. This provides a foundational infrastructure that can help the field of computational drug discovery grow in a healthy way.
📜Title: KG-Bench: Benchmarking Graph Neural Network Algorithms for Drug Repurposing 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.10.13.682003v1