
Table of Contents
- The RoFt-Mol benchmark shows there’s no single best way to fine-tune AI molecular models. The right strategy depends on task type and data size. A new method, DWiSE-FT, performs especially well with self-supervised pretraining.
- The QW-MTL framework combines quantum chemical descriptors with learnable task weights to offer a more accurate and efficient multi-task learning solution for predicting drug ADMET properties.
- Researchers trained an AI model, SurGBSA, on massive molecular dynamics simulation data. It predicts binding energies with high accuracy at nearly 6,500 times the speed, making physics-based calculations feasible for high-throughput screening.
- Handling pharmacokinetics (PK) data from complex tables is difficult. AutoPK is a new method that creates a standardized workflow for Large Language Models, improving data extraction accuracy and reliability so that small, open-source models can match commercial ones.
- SESAME is a generative AI model that efficiently predicts the conformational change of a protein from its unbound (apo) to its bound (holo) state, providing high-quality structures for virtual screening and cryptic pocket discovery.
1. RoFt-Mol Benchmark: What’s the Best Way to Fine-Tune AI Models for Drug Discovery?
In drug discovery, we often use large models, specifically Molecular Graph Foundation Models (MGFMs). These models are like brilliant graduates with a wealth of knowledge but no experience on our specific project. Our job is to “fine-tune” them so they can solve a particular problem, like predicting a molecule’s activity or toxicity.
But fine-tuning is tricky. If done wrong, the model might overfit the small dataset we provide and fail on new data. Or, it might “forget” its broad, pretrained knowledge and become shortsighted. It’s like teaching a Michelin-star chef to cook a family recipe—you want them to learn it without forgetting all their other culinary skills.
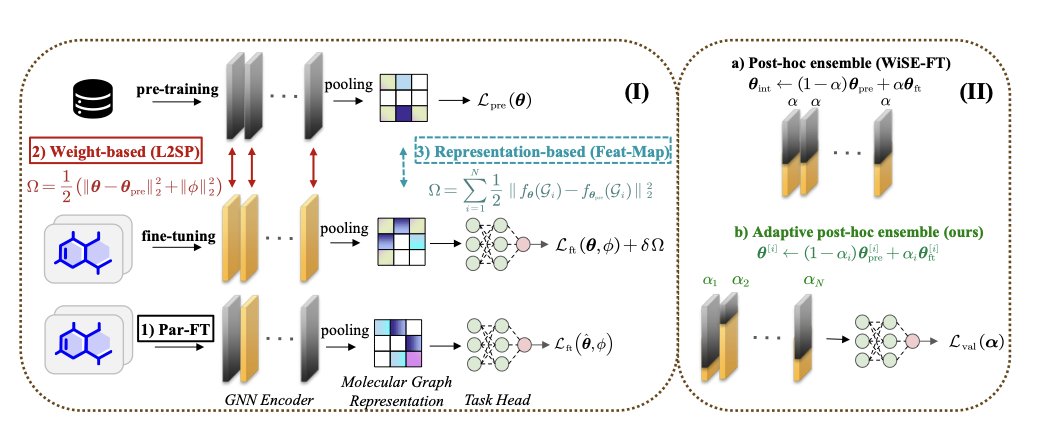
A new paper introduces the RoFt-Mol benchmark to address this problem. The researchers built a standardized testbed to systematically evaluate eight common fine-tuning methods, showing us which one to use in which situation.
First, consider the model’s pretraining: supervised or self-supervised?
A model’s pretraining method directly affects how it responds to fine-tuning.
Supervised pretraining is like specialized training. The model is trained on specific tasks, such as predicting molecular properties. Self-supervised pretraining is more like a general education, where the model learns by studying the rules of molecular structure itself.
The study found that in few-shot scenarios, where we have only a small amount of labeled data, supervised models perform better. This makes sense; the specialist can apply its knowledge directly.
But with enough data, the situation changes. The advantage of a supervised model only holds if its pretraining task is highly relevant to the new fine-tuning task. Otherwise, a self-supervised model, with its more solid foundation, may perform better.
Next, consider the task type: classification or regression?
Our tasks generally fall into two categories:
- Classification: Answering “yes” or “no.” For example, is this molecule “active” or “inactive”?
- Regression: Predicting a specific numerical value. For example, what is the exact solubility of this molecule?
The paper found that models are more prone to overfitting on classification tasks, especially with small datasets. This is because the model can just memorize the labels for a few samples. Regression tasks, however, require the model to understand the precise relationship between input and output to predict a number, forcing it to learn the underlying patterns.
This is a reminder for our daily work: be extra cautious about overfitting when dealing with small-sample classification problems.
Which fine-tuning methods work best?
The RoFt-Mol benchmark tested eight methods across three categories. Let’s focus on the key ones.
Weight-based methods directly adjust the parameters of the pretrained model.
These two methods are particularly effective when paired with self-supervised pretrained models.
Representation-based methods freeze most of the pretrained model’s parameters and only train a few newly added layers.
DWiSE-FT: A stronger all-rounder
Finally, the researchers developed a new method called DWiSE-FT. It combines the advantages of WiSE-FT and L2-SP, allowing it to balance old and new knowledge like WiSE-FT while preventing the model from straying too far from its original parameters, like L2-SP.
Results show that DWiSE-FT performs well on self-supervised pretrained models, delivering robust performance on both classification and regression tasks. This is good news for researchers who need a reliable, versatile tool for different projects.
The RoFt-Mol benchmark gives us a practical guide for fine-tuning. The next time you choose a fine-tuning strategy, you can make a more informed decision based on the model’s pretraining, the task type, and the amount of data you have.
📜Title: RoFt-Mol: Benchmarking Robust Fine-Tuning with Molecular Graph Foundation Models 📜Paper: https://arxiv.org/abs/2509.00614
2. Quantum-Enhanced ADMET Prediction: A More Accurate and Faster AI Framework
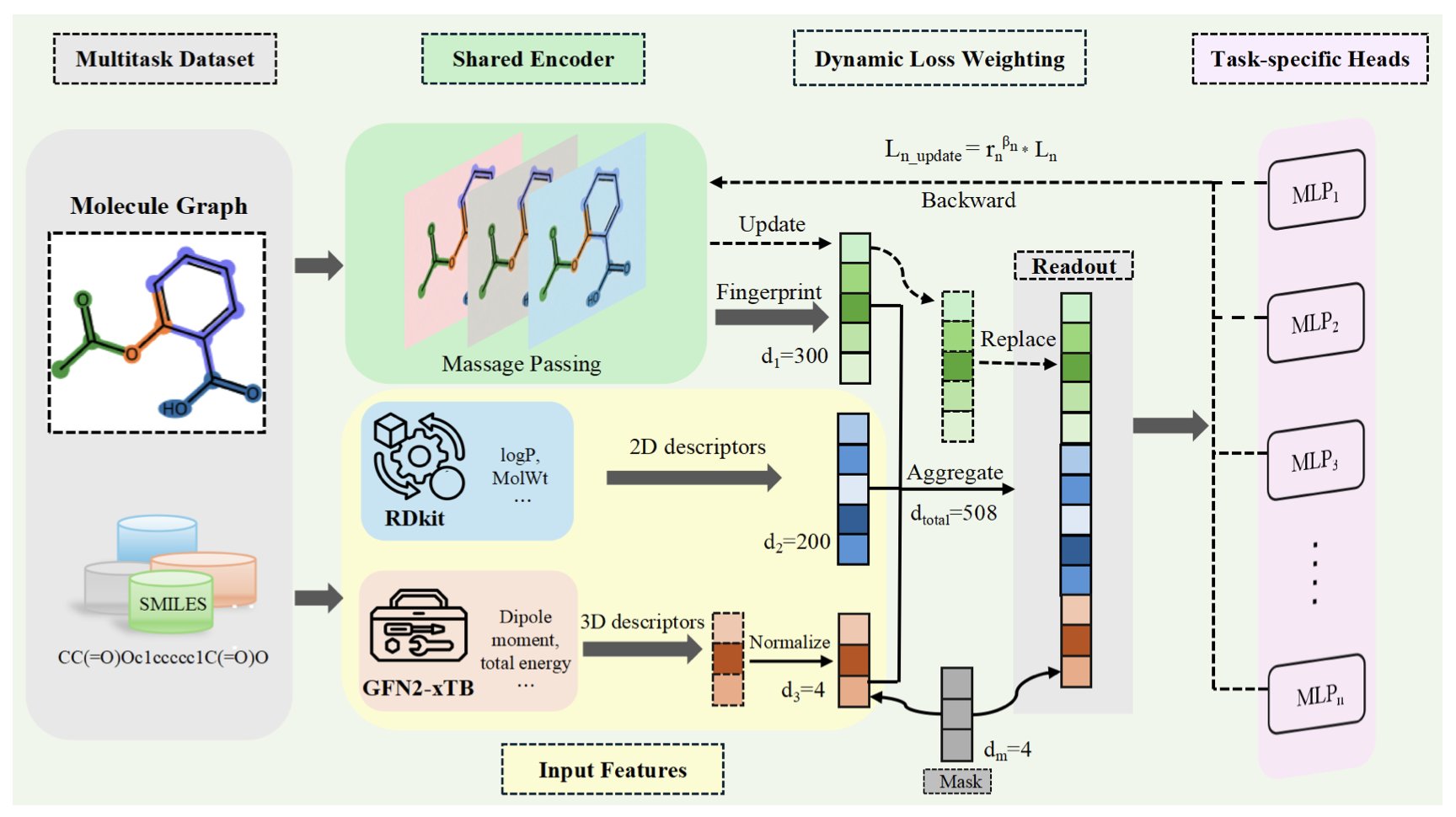
In early-stage drug discovery, quickly and accurately predicting a candidate molecule’s ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties is a core challenge. Prediction failures are a major reason for late-stage clinical trial failures. Multi-Task Learning (MTL) is a natural approach, where one model learns multiple related properties at once, like predicting both solubility and liver toxicity. This helps the model learn more general molecular features. But it has a problem: data imbalance. There might be hundreds of thousands of data points for solubility but only a few thousand for a specific toxicity. The model will naturally favor the task with more data, leading to poor performance on smaller tasks.
The QW-MTL framework proposed in this paper aims to solve this.
First, it improves the molecular representations. Traditional molecular fingerprints like ECFP describe a molecule’s topological structure. But many ADMET properties, such as metabolic stability and membrane permeability, are closely tied to a molecule’s electronic properties. The authors combined descriptors from quantum chemistry calculations (like frontier orbital energies and dipole moments) with traditional descriptors. This is like giving the model not just a “skeleton” of the molecule, but also a map of its “electron cloud.” With these physically meaningful features, the model gains a deeper understanding of the molecule and makes better predictions.
Second, and the core innovation of the framework, is its “learnable task weighting” mechanism. The authors designed a strategy that allows the model to dynamically adjust the weight of each task during training based on its sample size. Tasks with fewer samples are given higher weights, forcing the model to focus more on learning them. This is like an experienced tutor who spends more time on a student’s weak spots instead of repeating what they already know. This simple mechanism effectively addresses the issue of task imbalance in multi-task learning.
The results show this approach works. The researchers tested QW-MTL on 13 public benchmark datasets from the Therapeutics Data Commons (TDC). Its performance surpassed traditional single-task learning models and achieved state-of-the-art results on several tasks.
It is also practical. Adding quantum features and dynamic weighting did not significantly increase the number of model parameters; in fact, inference speed was faster. This allows it to be integrated into existing high-throughput virtual screening pipelines. For a drug discovery team, a model that is both accurate and fast means screening millions of molecules at a lower cost and in less time, increasing the probability of finding good drug candidates.
📜Title: Quantum-Enhanced Multi-Task Learning with Learnable Weighting for Pharmacokinetic and Toxicity Prediction 📜Paper: https://arxiv.org/abs/2509.04601
3. SurGBSA: AI Speeds Up Binding Energy Prediction by Nearly 6,500x
In drug discovery, we are always trading off speed and accuracy. Molecular docking is fast but often inaccurate. Methods like MM/GBSA, which calculate binding free energy through Molecular Dynamics (MD) simulations, are more reliable but prohibitively slow. You can’t use them for virtual screening campaigns that need to evaluate millions of compounds. It’s like trying to find a needle in a haystack with a high-powered microscope—theoretically possible, but the project would be over by the time you found it.
This work introduces SurGBSA to solve this problem. Since MM/GBSA is so slow, can we train an AI model to learn its predictions and then make those predictions at AI speeds?
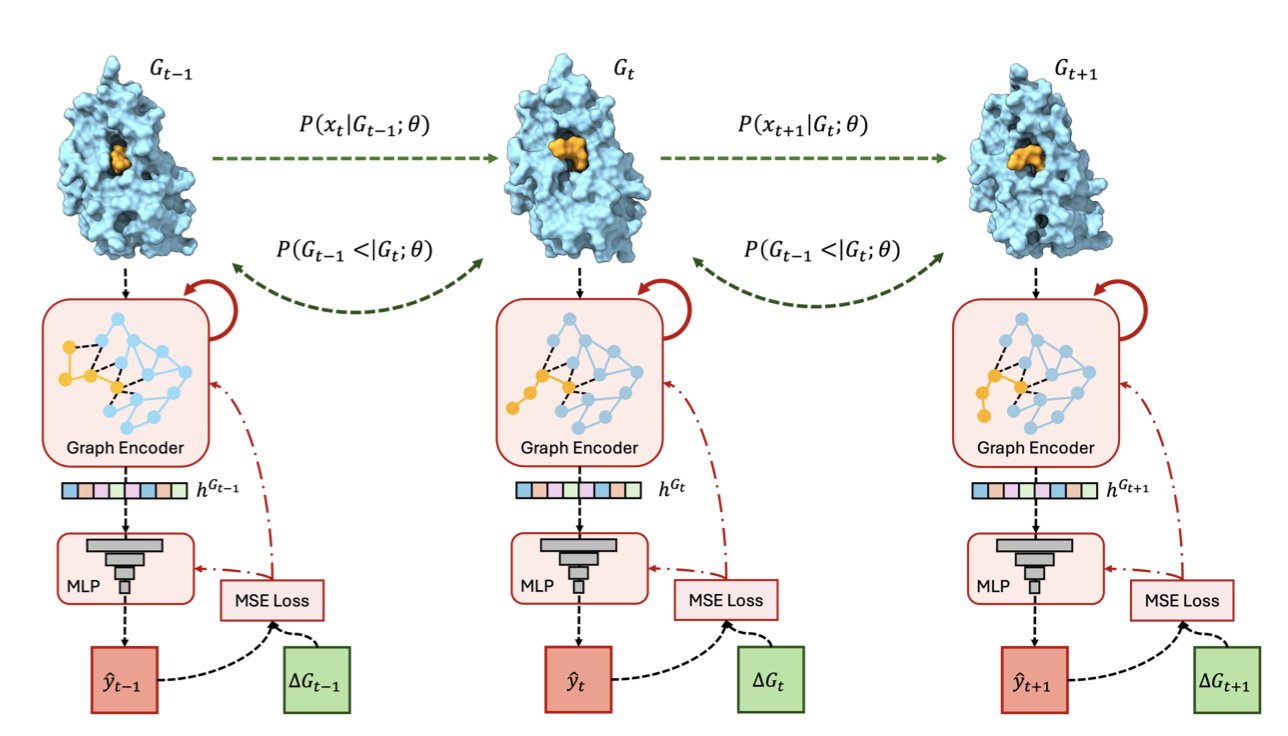
The key to success: Training AI with “movies” instead of “photos”
Traditional machine learning models often use static 3D structures, like crystal structures, as input. This is like showing the AI a single “photo” of a compound bound to a target; the information is limited. In reality, molecules and proteins are constantly moving and vibrating, their conformations always changing.
The creators of SurGBSA took a different approach. They first ran extensive MD simulations to generate over 1.4 million dynamic “movie clips” (3D trajectories) of molecule-protein complexes. Then, they trained their model using these information-rich movies. The benefit is that the model learns not just a single static conformation but the full range of conformations and energy changes that occur during an interaction. This makes the model’s generalizations more robust.
A surrogate model that’s nearly 6,500 times faster
SurGBSA is a surrogate model. This means the researchers have already done the most time-consuming work for us: generating massive amounts of MD data and training the model.
Now, our workflow changes. When we get a docked pose, we no longer need to run a lengthy MD simulation for it. We just feed the pose to the SurGBSA model, and it instantly predicts the MM/GBSA energy value. According to the paper, this process is 6,497 times faster than the traditional calculation. A computation that once took hours or even days now takes seconds.
This speed does not come at a great cost to accuracy. In the task of ranking binding poses, SurGBSA’s predictions were highly correlated with traditional MM/GBSA calculations (Pearson correlation of 0.702). This means it can effectively distinguish good binding modes from bad ones.
What this means for the industry
SurGBSA makes it possible to incorporate more reliable physicochemical calculations early in high-throughput screening. We can use a scoring function with near-MM/GBSA accuracy to screen millions of molecules, which should increase hit rates and reduce downstream experimental costs.
The researchers also compared different graph neural network architectures (GNN, EGNN, EGMN) and found that a pretrained EGMN performed best. This shows that leveraging existing knowledge through pretraining can further improve model performance. They have made their code and data public, which not only demonstrates their confidence but also paves the way for developing the next generation of MD-based foundation models.
📜Title: SurGBSA: Learning Representations From Molecular Dynamics Simulations 📜Paper: https://arxiv.org/abs/2509.03084v1
4. AutoPK: Using Large Models to Accurately Extract Pharmacokinetics Data
Drug discovery researchers frequently need to extract pharmacokinetics (PK) data from a vast body of literature. This data is often buried in PDF tables with inconsistent formatting and terminology. For instance, “half-life” might be recorded as Half-life, T1/2, or t_half. Manually curating this data is time-consuming and error-prone.
Asking Large Language Models (LLMs) to process these complex tables directly does not work well. The models often misinterpret data or invent it, a phenomenon known as “hallucination.”
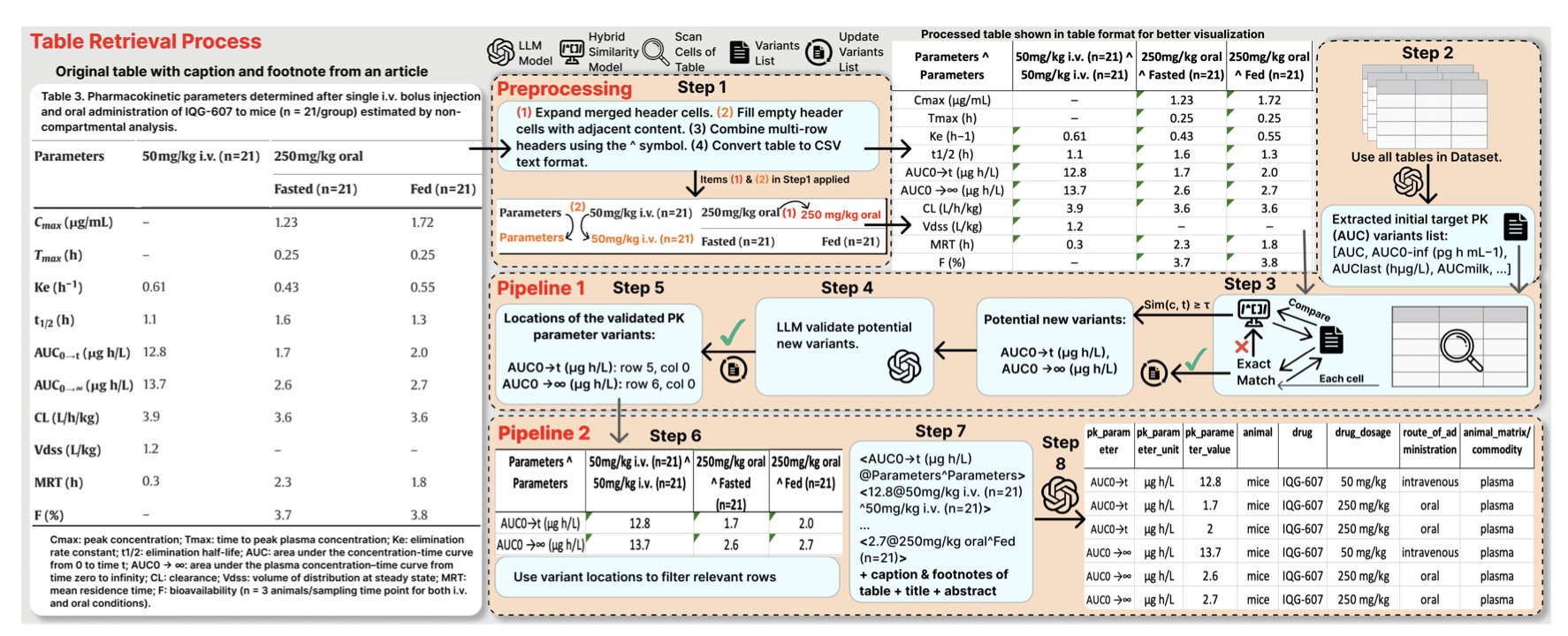
AutoPK solves this problem with a two-stage strategy.
The first step is identification and standardization. AutoPK scans a table to find all potential PK parameter terms. It uses a hybrid similarity metric, combining lexical form (like CL and Clearance) and semantics (both refer to “clearance”), to map different terms to a standard vocabulary. For example, T1/2 and t_half would both be standardized to Half-life.
The second step is extraction and reconstruction. After standardizing the terms, AutoPK converts the table into a simple key-value text format, such as {'Half-life': '2.5 h', 'Species': 'Rat'}. Finally, the LLM reads this clean, standardized text to generate a structured, machine-readable table.
This process is like giving the LLM a professional prep assistant. The assistant first cleans and prepares the raw ingredients (the original table) before handing them to the chef (the LLM) to cook (generate the final output). This reduces the chance of errors.
On a dataset of 605 real-world PK tables, the LLaMA 3.1-70B model, assisted by AutoPK, improved its F1-scores for half-life and clearance parameters by 0.10 and 0.21, respectively, reaching final scores above 0.9.
AutoPK’s impact on smaller models is particularly significant. When processing data directly, models like Gemma 3-27B and Phi 3-12B had hallucination rates as high as 60-95%. With the AutoPK workflow, their hallucination rates dropped to 8-14%, and their F1-scores increased by 2 to 7 times.
This means that with AutoPK’s help, the open-source Gemma 3-27B model can extract certain PK parameters with performance that surpasses the commercial GPT-4o Mini model. This offers drug discovery organizations a low-cost, high-efficiency, and reliable data extraction solution.
AutoPK’s value lies in showing how to use existing tools more cleverly, rather than simply pursuing larger models. It addresses a specific and common industry problem, advancing the automation and scaling of drug data analysis.
📜Title: AutoPK: Leveraging LLMs and a Hybrid Similarity Metric for Advanced Retrieval of Pharmacokinetic Data from Complex Tables and Documents 🌐Paper: https://arxiv.org/abs/2510.00039v1
5. The SESAME Model: AI Predicts Protein Conformations to Open Cryptic Drug Pockets
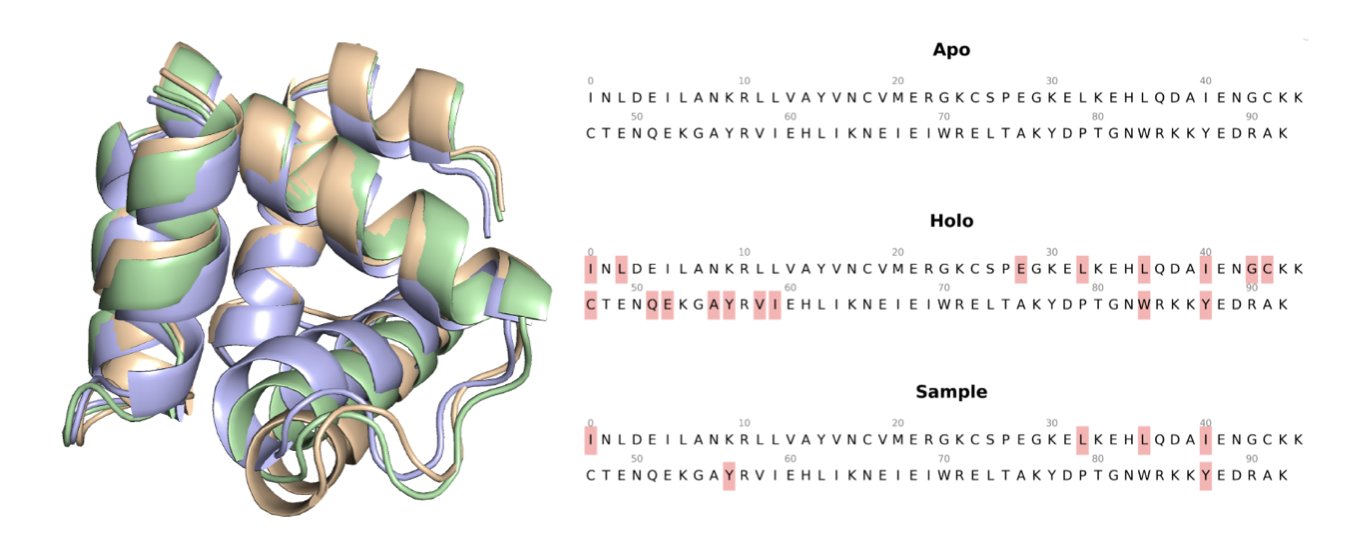
In drug discovery, proteins are not static blocks but dynamic machines. The structures we get from crystal structure databases like the PDB are typically the apo conformation, which is the protein’s “unoccupied” state. But a drug molecule binds to the holo conformation, its “in-use” state. The two can be very different, especially around the binding pocket.
Traditionally, simulating this conformational change requires Molecular Dynamics (MD). This method is powerful but slow and computationally expensive. A single MD simulation can take days or weeks, making it impractical for early-stage discovery that involves screening millions of compounds.
The SESAME model offers an alternative. It is a generative AI trained specifically to learn how a protein “morphs” from its apo state to a holo state.
It works using flow matching. You can think of it as learning a transformation rule. The model takes an apo conformation as input and then guides the structure, as if in a current of water, until it “flows” into a holo conformation that is more likely to bind a ligand. This process is handled mathematically in SE(3) space (for rotation and translation) and R³ space (for atomic coordinates) to ensure the generated conformations are physically plausible.
Researchers tested it on the D3PM-Large dataset. In 38% of predictions, the Root Mean Square Deviation (RMSD) between the generated structure and the true holo structure was less than 2.0 Å. In structural biology, 2 Å is a key threshold for accuracy; a value below this generally means the predicted backbone structure is close to the real one.
One of the most exciting aspects of this model is its ability to help find “cryptic pockets.” These are pockets that are normally closed or non-existent and are only exposed transiently as the protein moves. They are often excellent drug targets because they can be more selective, but they are hard to find using static structures. The researchers combined SESAME with another pocket-finding tool, PocketMiner, and found that the dynamic conformations generated by SESAME increased the success rate of discovering cryptic pockets.
Ultimately, the generated structures must be useful for drug design. The researchers performed molecular docking tests with them. The results were clear: using the holo conformations generated by SESAME led to better docking scores and higher success rates than using the original apo structures. This means we can integrate it into virtual screening workflows. We can first use SESAME to quickly generate a set of high-quality protein conformations and then perform large-scale compound screening, which should improve hit rates.
Of course, the model has room for improvement. It currently focuses on the protein backbone. The next step is to incorporate the fine-grained conformations of side chains, as the interactions between a drug and amino acid side chains are key to binding.
📜Title: SESAME: OPENING THE DOOR TO PROTEIN POCKETS 📜Paper: https://arxiv.org/abs/2509.05302