
Table of Contents
- By combining knowledge of protein structure and function, AI can more accurately predict the pathogenicity of hemoglobin mutations.
- This explainable AI model precisely predicts compound water solubility and quantifies the impact of classic physicochemical parameters like LogP and TPSA, giving medicinal chemists specific directions for optimization.
- The SynGA algorithm doesn’t generate molecular structures directly. Instead, it performs genetic operations on a molecule’s synthesis route, ensuring every designed molecule has a clear synthesis method.
1. AI Maps Hemoglobin Mutations to Precisely Predict Genetic Diseases
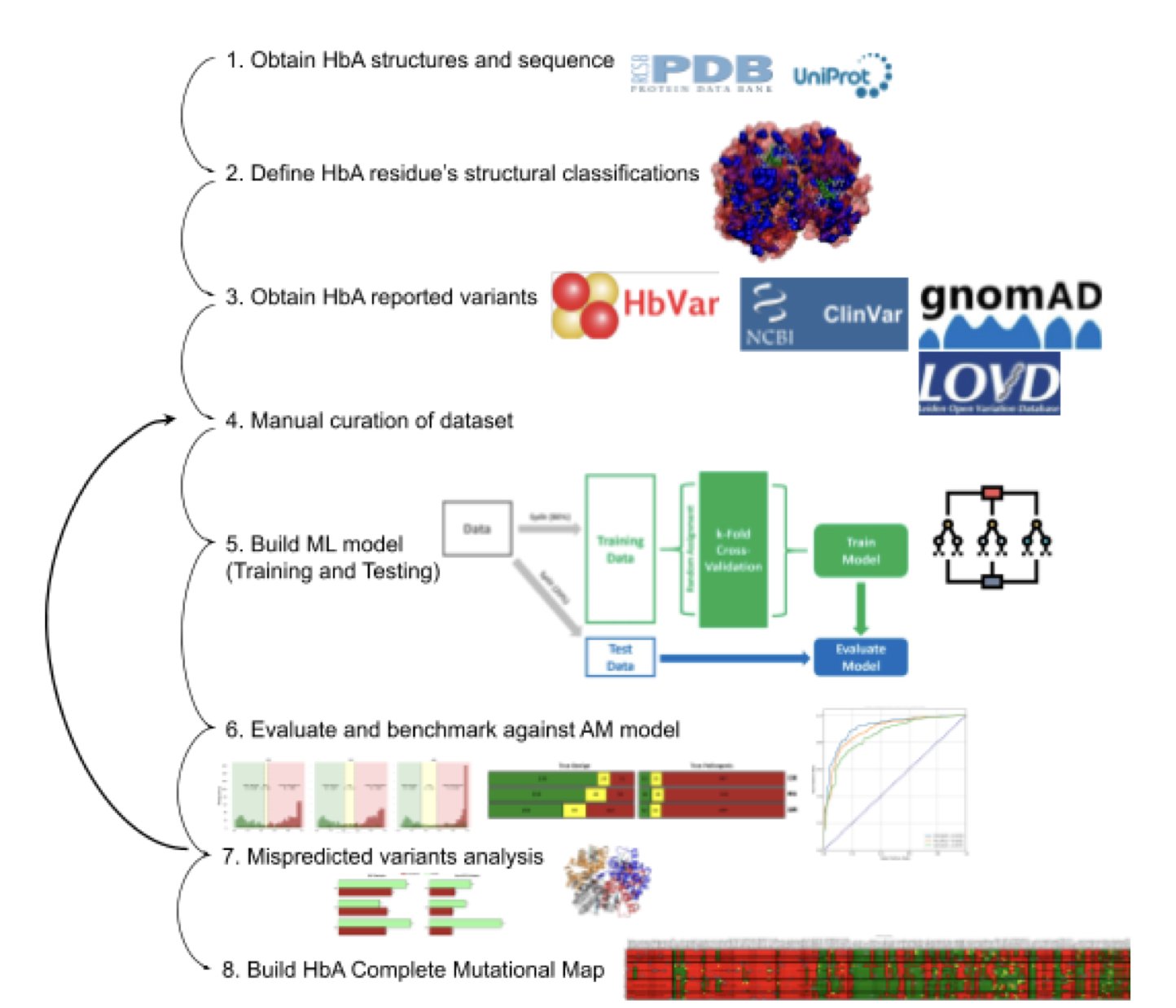
Determining whether a Single Amino Acid Substitution (SAS) in a gene sequence is harmless or pathogenic is a tough problem in drug development and clinical diagnosis. This is true even for a well-studied protein like human hemoglobin (HbA). Hemoglobin transports oxygen in the blood, and small malfunctions can lead to sickle cell disease or thalassemia.
When doctors find a new hemoglobin mutation, they often classify it as a “Variant of Uncertain Significance” (VUS). This classification doesn’t confirm a diagnosis or rule out risk. One research team tried to solve this problem with AI.
Instead of having AI learn directly from massive amounts of sequence data, they first integrated decades of “expert knowledge” on hemoglobin.
First, the team built a high-quality dataset. It included the pathogenic classifications of known mutations and also annotated each one with rich structural and functional information. For example, is the mutation site inside the protein or on its surface? Does it affect protein stability? Is it in a key functional area for oxygen binding?
Second, the team built a predictive model. They didn’t rely solely on a Large Language Model like AlphaMissense. AlphaMissense can predict the probability that a mutation is harmful by comparing protein sequences across many species, but it’s like a generalist with broad knowledge, lacking deep understanding of a specific protein. The researchers combined AlphaMissense’s prediction scores with the structural and functional features they had compiled to train a new model. This was like giving the generalist an expert consultant, combining breadth with depth.
This combined model had a higher prediction accuracy. It also successfully reclassified many “Variants of Uncertain Significance” as either likely pathogenic or likely benign, providing clearer guidance for clinical diagnosis.
Finally, the team used the optimized model to predict the effect of all 5,453 possible single amino acid mutations in hemoglobin, creating a complete functional map of mutations.
This map is like a high-resolution chart, showing “safe zones” (where mutations are tolerated) and “danger zones” (where mutations could cause functional problems). For instance, regions in the protein’s core that maintain structural stability and the pocket that binds to heme are extremely sensitive to mutations. This aligns with existing knowledge of protein structure and function.
This work shows that combining classic structural biology and biochemistry knowledge with machine learning models can improve predictive performance. The method also serves as a model for predicting the effects of variants in other proteins.
📜Title: A Structure and Function-Based Complete Mutational Map of Human Hemoglobin Using AI 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.09.30.679504v2
2. AI Quantifies Drug Solubility: LogP is Key, but the Details Matter
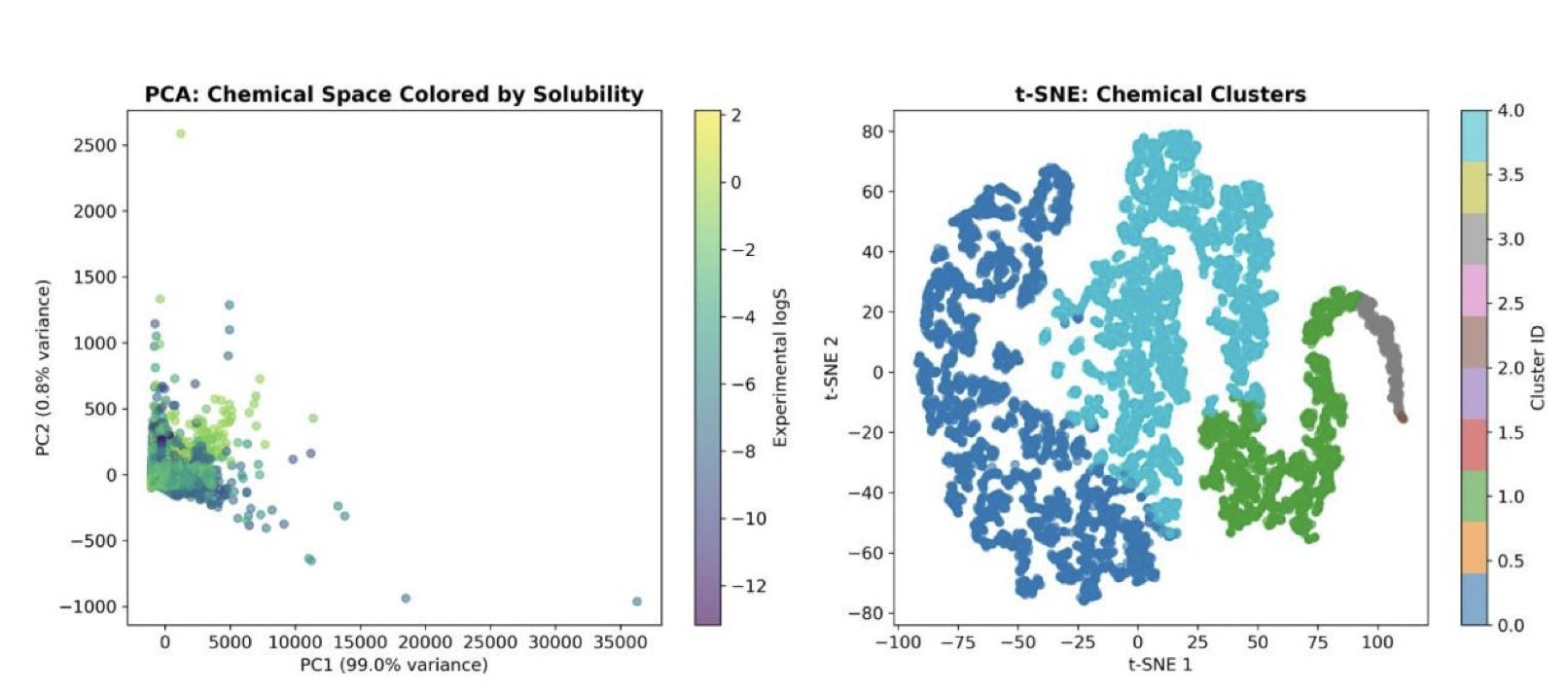
Water solubility is a central challenge in drug discovery. Medicinal chemists follow rules of thumb like the “Rule of Five” to avoid making molecules too hydrophobic. But these rules are qualitative. For example, lowering the octanol-water partition coefficient (LogP) can improve solubility, but the specific thresholds and the influence of different functional groups often depend on intuition and trial and error.
Work by Ali Afshari’s team uses machine learning to quantify this chemical intuition. The highlight is the model’s interpretability, which reveals why certain factors affect solubility.
The team used a dataset of over 9,000 compounds to train a LightGBM model, a type of gradient boosting decision tree. The model’s predictions were accurate, achieving a coefficient of determination (R²) of 0.80. The core value of the work, however, lies in the analysis that followed.
The team used the SHAP (SHapley Additive exPlanations) method. SHAP is an attribution tool that can calculate the specific contribution of each molecular feature (like LogP or TPSA) to every prediction.
The analysis confirmed chemical intuitions with concrete numbers: 1. LogP plays the dominant role. This parameter contributed nearly 40% of the predictive weight. Hydrophobicity is the fundamental factor determining if a molecule will dissolve in water. For a hydrophobic molecule to dissolve, energy is needed to break the existing hydrogen bond network between water molecules. 2. TPSA has the next largest impact. TPSA, or topological polar surface area, measures the total surface area of polar atoms in a molecule, which directly relates to its ability to form hydrogen bonds with water. The larger the polar surface area, the more easily the molecule can interact with water.
The model also uncovered more subtle relationships.
The model’s practical value is its ability to provide local explanations for individual molecules. For example, it can quantify the positive contribution of a specific carboxyl group to solubility or the negative impact of an alkyl chain. This analysis makes molecular optimization more specific, moving from a general principle like “lower the LogP” to a concrete strategy like “modify this specific group to efficiently lower the LogP.”
This research is a great example of combining machine learning with physical chemistry. It goes beyond black-box predictions to look inside the model, translating its decision logic into language chemists can understand and use. This turns AI into a tool that aids both thought and design.
📜Title: Beyond Prediction: Mechanistic Elucidation of Molecular Drivers for Aqueous Solubility Using Interpretable Machine Learning 🌐Paper: https://doi.org/10.26434/chemrxiv-2025-xq8v3
3. SynGA: Starting from the Synthesis Route to Avoid Unmakeable Molecules
A common problem in computer-aided drug discovery is that molecules designed by AI, even with great theoretical activity, may be impossible to synthesize in a lab. This kind of design wastes both time and computing resources.
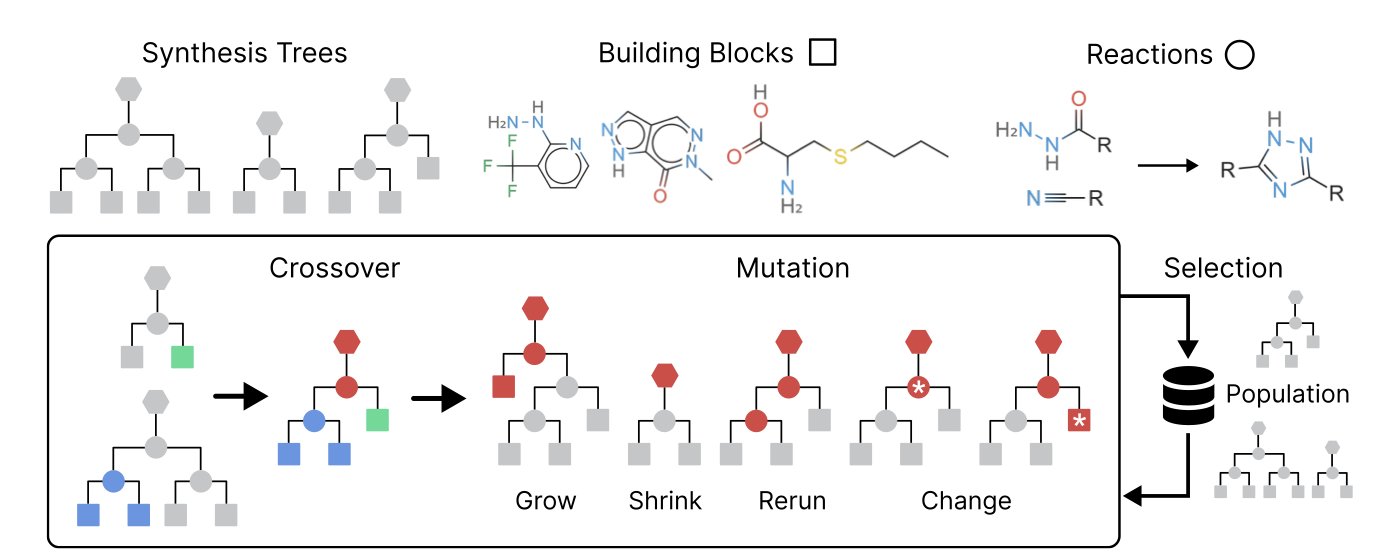
The SynGA algorithm tries to solve this problem. Traditional algorithms first generate a molecular structure and then try to predict its synthesizability. SynGA’s approach is the reverse: it doesn’t operate on molecules directly, but on their synthesis routes.
SynGA treats a synthesis route like a gene and uses a Genetic Algorithm to evolve it. The algorithm has two basic operations:
- Crossover: Randomly select two valid synthesis routes and swap parts of them to create a new, valid route.
- Mutation: Randomly change a step in a route, for example by swapping a building block or altering reaction conditions.
By repeating crossover and mutation, the algorithm explores a “synthesizable chemical space” made up of known chemical reactions and building blocks. Since every operation is based on real chemistry, any new route produced corresponds to a molecule that is guaranteed to be synthesizable. Synthesizability is therefore a built-in constraint of the algorithm, not a property checked afterward.
To make the search more efficient, the researchers added a machine learning filter. This filter pre-evaluates and screens building blocks, prioritizing starting materials that are more likely to produce the target molecule.
When SynGA is integrated into a Bayesian Optimization framework, it becomes SynGBO. This version is designed for goal-oriented tasks, such as finding the molecule that binds most strongly to a specific protein target. Bayesian optimization guides SynGA’s search, speeding up the process of finding the best solution in the chemical space.
The paper’s data shows that SynGA and SynGBO perform well on several benchmark tests, including finding analogs of known drugs and optimizing both 2D and 3D properties of molecules.
The method’s value lies in its simplicity and practicality. It is computationally inexpensive, its logic is clear, and it solves the synthesizability problem at the source. This means that candidate molecules designed by computational chemists can be handed directly to synthesis teams for validation, shortening the cycle from design to experiment. It can be used as a standalone molecule generator or as a module embedded in existing drug discovery workflows.
📜Title: A Genetic Algorithm for Navigating Synthesizable Molecular Spaces 🌐Paper: https://arxiv.org/abs/2509.20719v1