
Table of Contents
- The ALIGNED framework aligns experimental data with biological knowledge, improving the accuracy of genetic perturbation predictions. It can also correct and refine knowledge bases, moving beyond pure black-box models.
- BindFlow automates the complex process of absolute binding free energy calculation, making it free and easy to use. This provides a reliable tool for predicting affinity in drug development.
- Researchers used Free Energy Perturbation (FEP) to successfully predict drug resistance mutations in intrinsically disordered regions (IDRs), offering a new approach to designing more durable drugs.
1. ALIGNED: An AI that Predicts Genetic Perturbations by Reconciling Data and Knowledge
Drug development often faces a dilemma. On one hand, we have massive amounts of genetic perturbation data from experiments like CRISPR screens. On the other, we have decades of accumulated biological knowledge in databases like KEGG and Reactome pathway maps. Ideally, these two sources should support each other, but in reality, they often conflict.
A deep learning model driven purely by data might make predictions that seem accurate but don’t make biological sense. A model based only on a knowledge base might fail to explain new experimental findings because the knowledge is incomplete or wrong. So, which one should we trust?
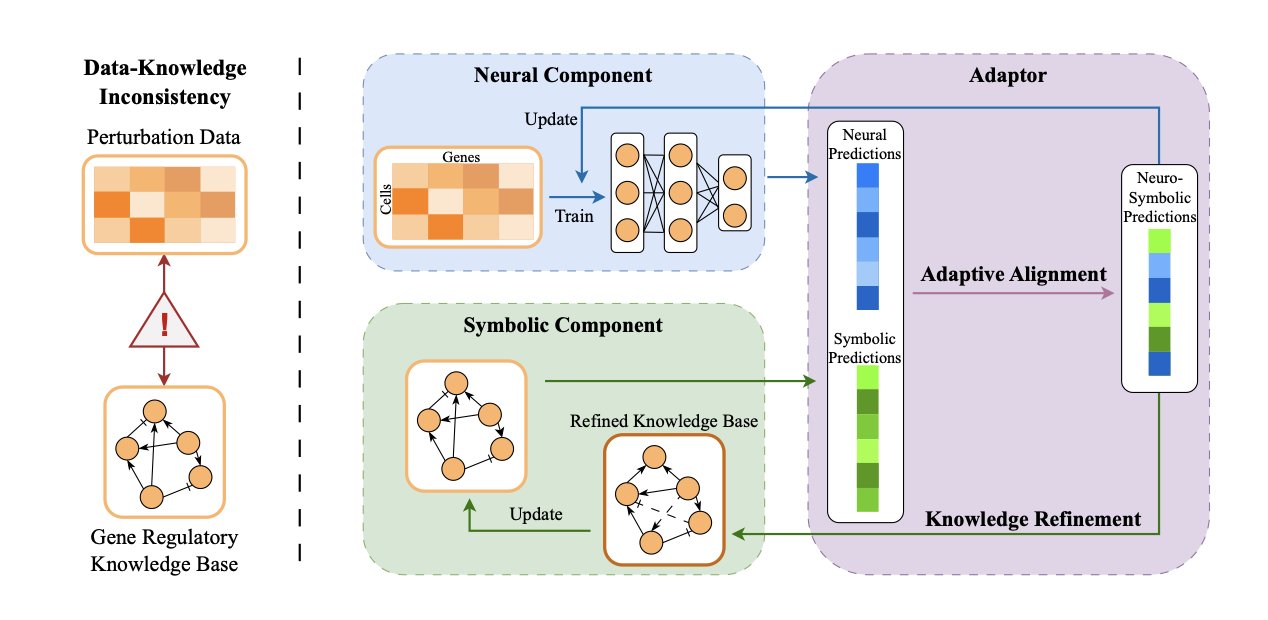
The ALIGNED framework, proposed in this paper, is designed to resolve this conflict.
Its core idea is Neuro-Symbolic Learning. Imagine a team with two roles. The “neuro” part is like an intuitive artist, a neural network that finds hidden patterns in messy data. The “symbolic” part is like a rigorous engineer who strictly follows known biological rules and pathway knowledge.
Previous methods listened to either the artist or the engineer. ALIGNED creates a communication channel between them. When data and knowledge disagree, it uses an adaptive alignment strategy to make a decision, rather than simply averaging the two. This process uses a gradient-free optimization mechanism that can flexibly handle both types of information.
To evaluate its decisions, the researchers designed a metric called “balanced consistency.” This metric requires predictions to both fit the experimental data and not significantly contradict the biological knowledge base. It’s like a clinical trial for a drug, where you need to see both efficacy (fitting the data) and a plausible mechanism of action (fitting the knowledge). This makes the results more trustworthy.
The framework can also update the knowledge base. If experimental data repeatedly points to a conclusion that contradicts the existing knowledge, ALIGNED can determine that the knowledge base may need an update. It uses gradient-based optimization and sparse regularization to correct regulatory relationships in the knowledge base. So, this model is not just a prediction tool; it’s a knowledge discovery engine. It can mine new biological pathways or molecular interactions from large datasets, helping our scientific knowledge evolve.
Tests on several large-scale datasets show that ALIGNED outperforms existing methods in both prediction accuracy and knowledge consistency. It performs especially well when data is limited, which is valuable for studying new targets where little data is available.
Work like ALIGNED represents a shift from pure black-box prediction toward intelligent systems that are interpretable, interactive, and can “talk” to domain knowledge. It uses the power of AI while also incorporating the knowledge accumulated by scientists.
📜Title: Adaptive Data-Knowledge Alignment in Genetic Perturbation Prediction 🌐Paper: https://arxiv.org/abs/2510.00512
2. BindFlow: Free, Automated FEP/MMPBSA for Drug Discovery
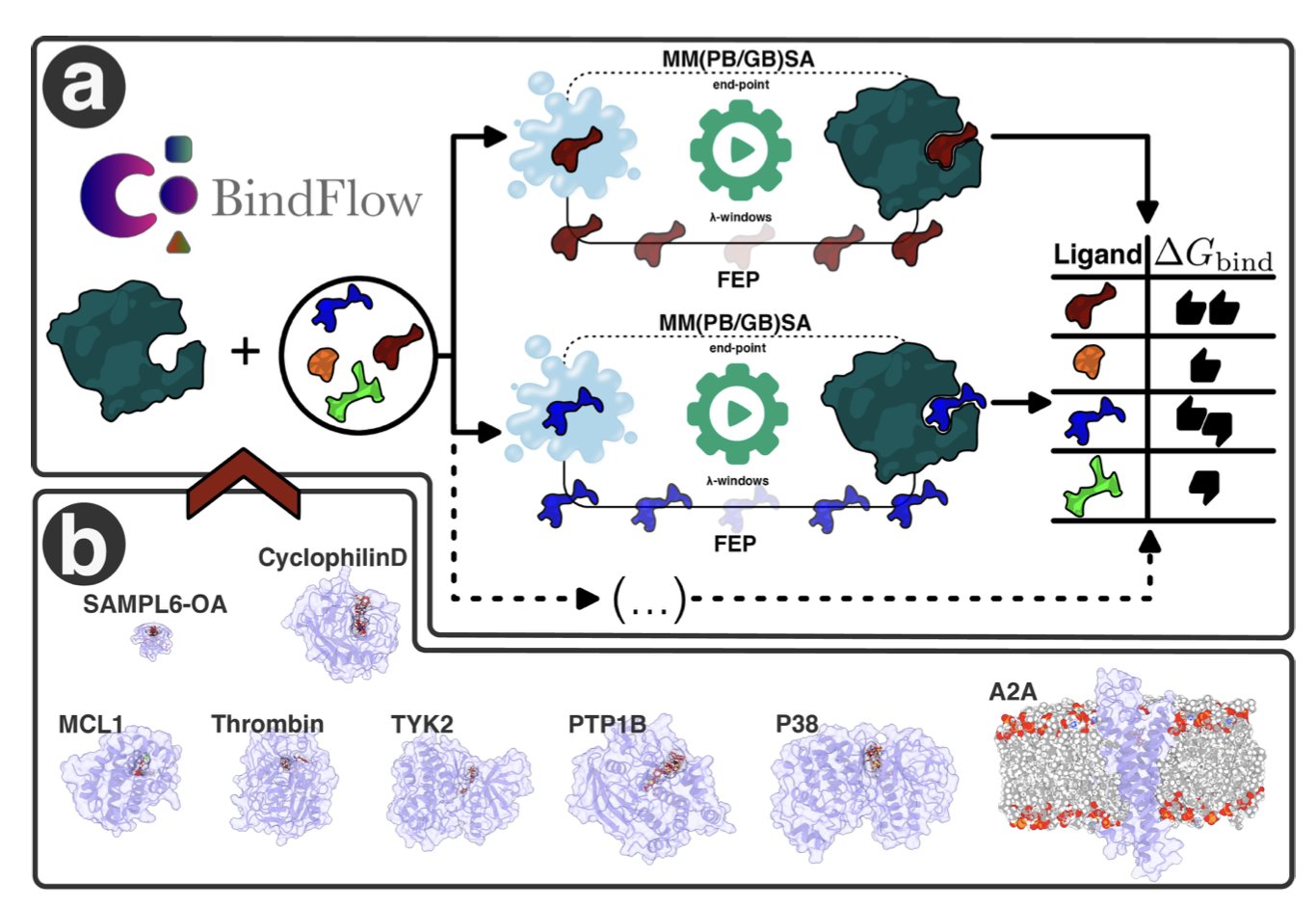
In drug discovery, accurately predicting the binding affinity of a small molecule to its target protein is a central challenge. Computational chemists have developed two types of tools for this. One type includes methods like Free Energy Perturbation (FEP), which are highly accurate but computationally expensive and labor-intensive. The other includes methods like MM(PB/GB)SA, which are fast but often considered less accurate.
For decades, using these tools required deep theoretical knowledge and tedious manual setup. High software costs and complex procedures also put them out of reach for many academic labs.
BindFlow is a tool designed to simplify this process. It’s a Python-based pipeline that automates all the steps for calculating Absolute Binding Free Energy (ABFE). Users just need to prepare the input files and run a command to get the results.
It’s free and open-source, so it avoids the high license fees of commercial software. It also supports multiple force fields, including GAFF, OpenFF, and Espaloma, giving users flexibility.
To validate the tool, the team tested it on a set of 139 ligand-target complexes. This set included soluble proteins, membrane proteins, and non-protein host-guest systems. The results showed that BindFlow’s calculations were comparable to industry standards, confirming the reliability of its data.
An important finding from the study relates to the MM(PB/GB)SA method. The conventional view is that end-point methods like MM(PB/GB)SA are less accurate than path-integral methods like FEP. But this study’s data showed that for certain systems and force field combinations, the affinity predicted by MM(PB/GB)SA can correlate with experimental values just as well as FEP.
This finding has practical implications. For instance, when screening a large virtual library for hit compounds, one could first use the less costly MM(PB/GB)SA for an initial pass. Then, computational resources could be focused on the most promising molecules for more precise calculation with FEP. This “coarse-to-fine” strategy balances efficiency and accuracy.
BindFlow turns a specialized computational task into a standardized, automated workflow. It improves the efficiency and reproducibility of binding free energy calculations, helping to speed up modern drug discovery.
📜Title: BindFlow: A Free, User-Friendly Pipeline for Absolute Binding Free Energy Calculations Using Free Energy Perturbation or MM(PB/GB)SA 🌐Paper: https://www.biorxiv.org/content/10.1101/2025.09.25.678545v1 💻Code: https://github.com/ale94mleon/BindFlow
3. FEP Tackles Flexible Proteins to Accurately Predict Drug Resistance
Some targets in drug discovery are hard to work with. Intrinsically Disordered Proteins (IDPs) and their Intrinsically Disordered Regions (IDRs) are a prime example. These regions lack a fixed 3D structure and are flexible like noodles, making structure-based drug design difficult. The N-terminus of the MDM2 protein is a classic IDR and a key target in the p53 tumor suppressor pathway.
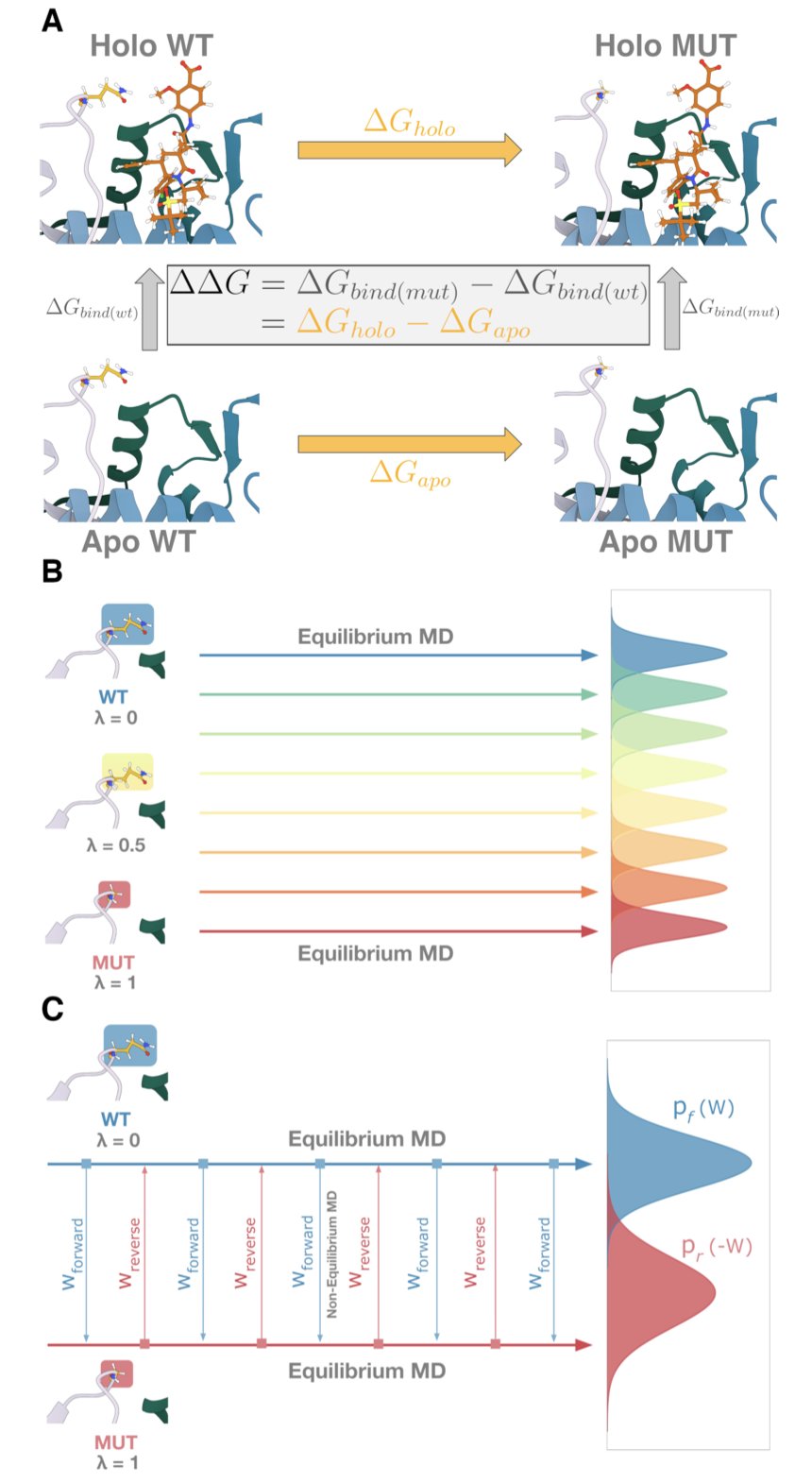
Drug resistance is another tough problem. A single amino acid mutation at the target site can render a drug ineffective. If we could predict which mutations cause resistance, we could design around them from the start. Free Energy Perturbation (FEP) is a computational method used to precisely calculate how mutations change binding energy. But applying it to dynamic IDRs has been a known challenge.
This work aimed to see if FEP could accurately predict drug resistance mutations for the IDR target MDM2.
The researchers compared two FEP calculation schemes: Equilibrium (EQ) and Non-equilibrium (NEQ). EQ-FEP allows the system enough time to adapt to a mutation and reach a new equilibrium. NEQ-FEP estimates the energy change by rapidly altering the system.
The results showed that EQ-FEP was more stable and accurate. Although its total computation time was longer, it needed only 10% of the sampling time to converge, with a final root-mean-square error (RMSE) of 1.48 kcal/mol. NEQ-FEP was faster, but its results were more variable, with an RMSE of 1.20 kcal/mol and a higher standard deviation. A stable, reproducible result is more valuable than a fast calculation that might only be accurate by chance.
The work also optimized the force field and water model. FEP calculations simulate the microscopic molecular world, and the force field and water model are the physical laws of that world. The researchers found that using the AMBER FF99SB*-ILDN force field with the OPC water model gave the best agreement with experimental data. For the I19G mutation, this combination reduced the RMSE for the AM-7209 complex from 0.79 kcal/mol to 0.39 kcal/mol.
This optimized protocol successfully predicted the binding affinity changes for two different ligands (AM-7209 and Nutlin-3a) with mutated MDM2, demonstrating the method’s general applicability.
This research extends the use of FEP as a predictive tool to the domain of IDRs. In the future, when developing drugs for flexible targets, we can predict resistance risk earlier and more accurately, which will help design next-generation drugs that are less likely to fail.
📜Title: Accurate Prediction of Drug Resistance for Intrinsically Disordered Protein Regions 🌐Paper: https://doi.org/10.26434/chemrxiv-2025-brzks