
AI in Drug Discovery: From SciGPT Beating GPT-4 to Trustworthy Drug Repurposing
This article explores three AI technologies for drug discovery and computational chemistry. SciGPT is a language model designed for scientific literature that outperforms GPT-4 on specific tasks. The REx model uses reinforcement learning to find explainable biological pathways for drug repurposing, addressing AI’s “black box” problem. And the strainedSMILES2xyz workflow solves an industry-wide challenge: reliably generating 3D structures for strained ring molecules from SMILES strings, ensuring the accuracy of computational simulations.
Contents
- SciGPT, optimized in architecture and training, is designed for scientific literature and outperforms the general-purpose LLM GPT-4 on specific tasks.
- REx uses a reinforcement learning reward system to find concise, biologically meaningful pathways in knowledge graphs, making AI recommendations for drug repurposing trustworthy.
- A new workflow combines existing tools to solve the problem of generating reliable 3D structures of strained molecules from SMILES strings.
1. SciGPT: An AI for Science That Performs Better Than GPT-4
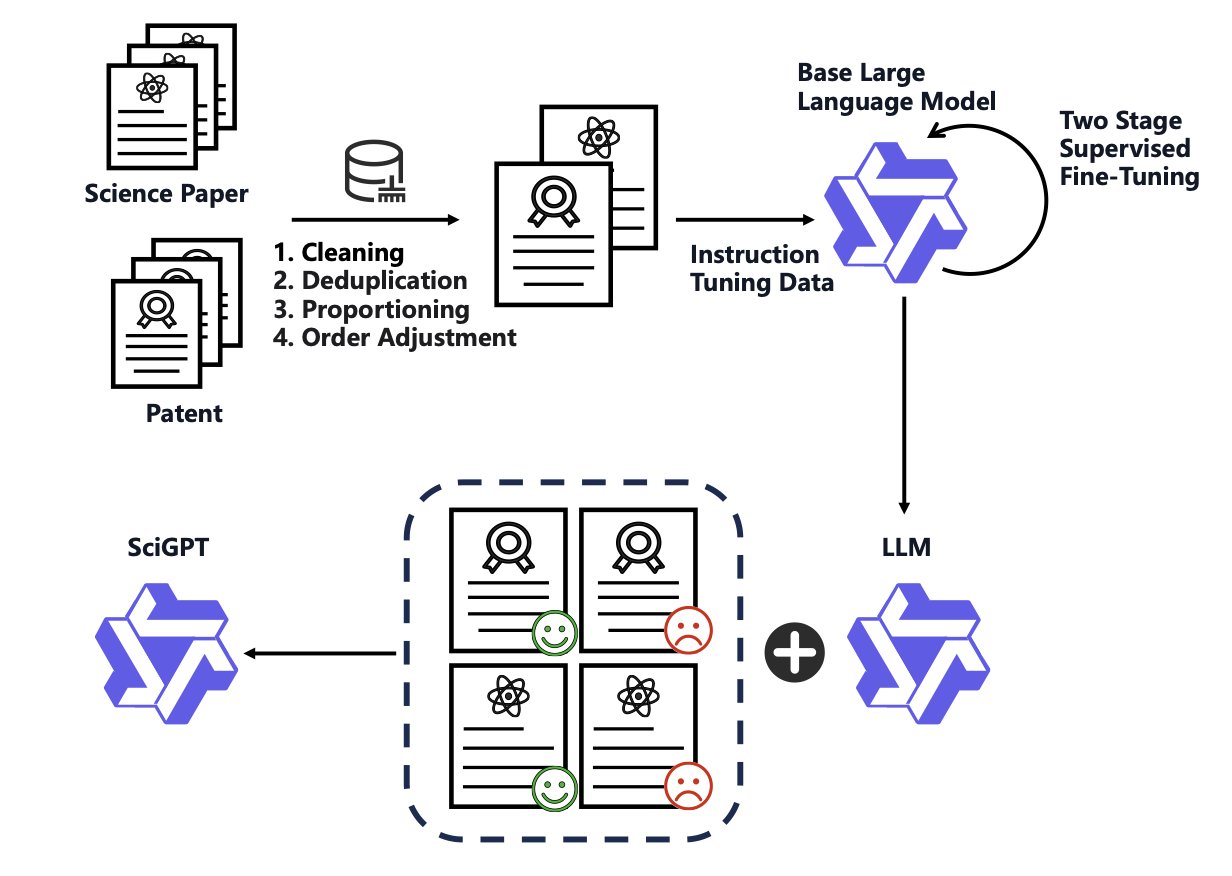
We’re all drowning in scientific papers. Keeping up with the latest progress in a field is a full-time job. General-purpose Large Language Models (LLMs) like GPT-4, while knowledgeable, struggle to read papers full of jargon and complex logic. It’s like asking a well-read humanities major to tackle a textbook on organic chemistry. They can understand the words, but they miss the deeper meaning.
SciGPT was built for this.
Think of GPT-4 as a knowledgeable generalist and SciGPT as a Ph.D. who has spent years in a specific field. It “gets” science better because of two key design choices.
First, it solves a practical engineering problem: memory. Processing a paper or technical report that’s dozens of pages long can exhaust the memory of many models. SciGPT uses a mechanism called a Sparse Mixture-of-Experts (SMoE). This is like having a committee of specialists. When a chemistry question comes up, the model activates its “chemistry expert” while the “physics” and “biology experts” rest. This uses only a fraction of the computational resources at any one time, lowering memory usage and making it easier to handle long documents.
Second, it uses a clever training method. The researchers used a two-step “distillation” approach. They first used the output of a larger model (the teacher) to train SciGPT (the student), allowing it to quickly grasp the basics of scientific language. Then, they fine-tuned it with high-quality scientific data. It’s like learning the fundamentals from a mentor and then tackling the latest papers on your own. They also integrated professional knowledge graphs (ontologies), which act like a built-in dictionary and relationship map. This helps the model understand connections between concepts, like the relationship between a specific kinase and a signaling pathway.
On ScienceBench, a benchmark designed for scientific tasks, SciGPT scored higher than GPT-4. This isn’t surprising. A general practitioner, no matter how knowledgeable, can’t compete with a specialist when diagnosing a rare genetic disease. Specialized models have an advantage on their home turf.
For people in drug discovery, this means future AI could more accurately extract compound structures, experimental data, and mechanisms of action from patents and papers. It might even spot hidden connections between different studies. For example, it could quickly screen the literature for all small molecules targeting KRAS G12C and summarize their ADME data. That saves us time.
The authors note that the next steps are to enable the model to process multimodal information like charts and graphs and to improve its explainability. In science, we want to know not just the answer, but also why. SciGPT is a good start.
📜Paper: https://arxiv.org/abs/2509.08032
2. REx Uses Reinforcement Learning to Provide Trustworthy Explanations for Drug Repurposing
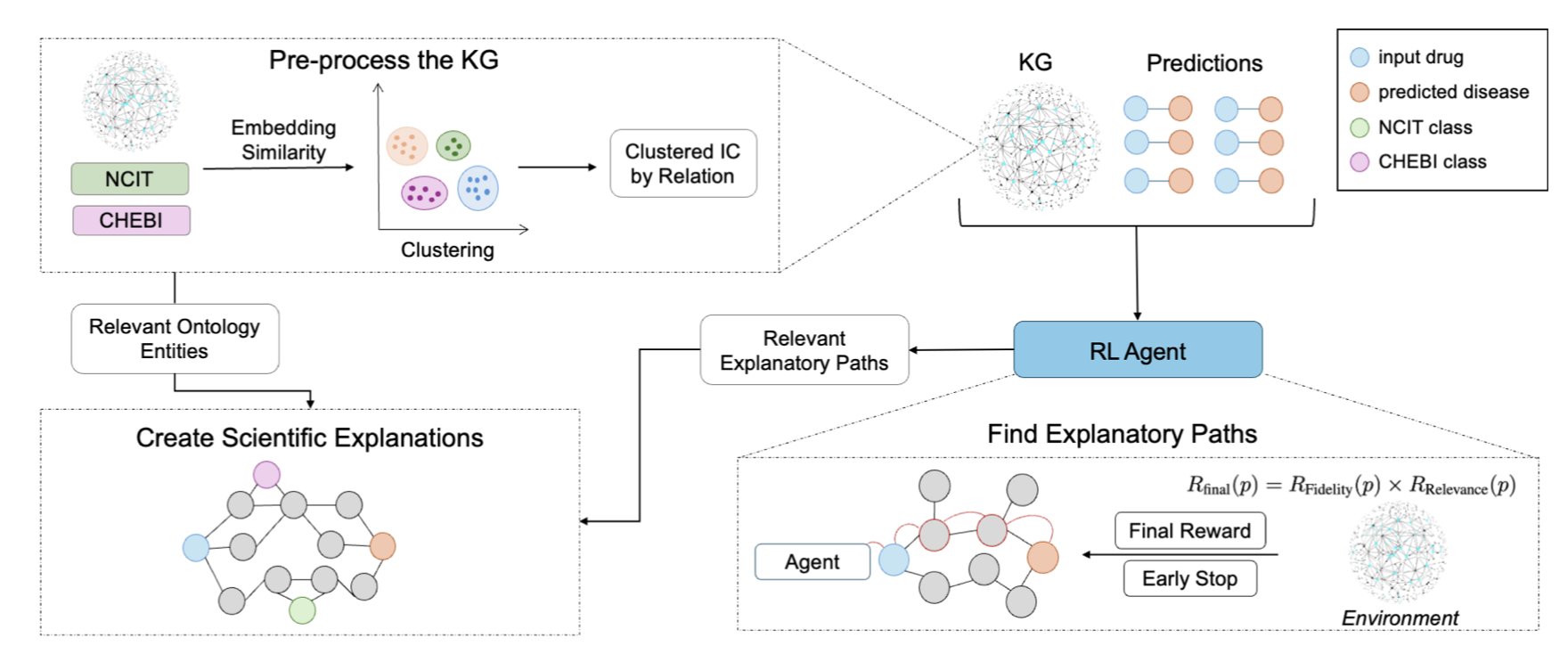
In drug development, we often hear that AI can recommend candidates for “drug repurposing.” But how does the AI come up with these ideas? Why should we trust its recommendations?
Most AI models are black boxes. You feed them data, they give you an answer, and you have no idea what happened in between. This is a big problem for drug development, which requires rigorous validation.
The REx model addresses this trust issue. It not only gives an answer but also clearly shows the reasoning path it took to get there. And that path has to make biological sense.
How REx Works
Imagine a massive biomedical Knowledge Graph (KG) as a city map. The map is covered with points, representing things like drugs, genes, diseases, and pathways. Roads connect these points, representing relationships, such as “Drug A inhibits Protein B,” “Protein B is involved in Pathway C,” and “Pathway C is related to Disease D.”
The task of drug repurposing is to find a path on this map from “Drug A” to “Disease D.”
A traditional model might find a path, but it could be a winding, convoluted route through many irrelevant nodes, making it meaningless. REx is different. It uses reinforcement learning to train an “agent” to find its way through the graph. The key is its reward system, which acts like a set of navigation rules for the agent.
Rule One: Take the shortcut, not the scenic route. REx introduces an “early termination” mechanism. If the agent’s path through the graph becomes too long with too many detours, it gets penalized. The simple idea here is that in biology, the most direct explanation is often the most likely one. This pushes the model to find more concise, core causal chains.
Rule Two: Take the main roads, not the back alleys. A short path isn’t enough; the path itself has to be meaningful. REx calculates the “Information Content” of each node and relationship along a path. A node or relationship that is more specific and important within the knowledge graph has a higher information content. For example, a path that goes through a well-known cancer pathway like p53 would score much higher than one that passes through an obscure metabolic enzyme. This ensures that the AI’s explanation is built on a solid foundation of biological knowledge.
How well does it work?
The researchers tested REx on three major biomedical knowledge graphs: Hetionet, PrimeKG, and OREGANO. The results showed that it not only surpassed current methods in prediction accuracy but, more importantly, generated higher-quality explanation paths.
They conducted an ablation study, where they removed key components of REx and tested it again. They found that taking out either the “early termination” or “information content reward” mechanisms caused a significant drop in performance. This proves that this reward system is central to REx’s success.
The most convincing evidence came from domain experts. The researchers showed the explanation paths generated by REx and other models to biomedical specialists and asked them to score them. The experts consistently rated the explanations from REx as more plausible and trustworthy.
AI in drug discovery shouldn’t just be a black box that spits out answers. It should be a research partner that can converse with scientists and provide testable hypotheses. The path REx generates, “Drug A -> Target B -> Pathway C -> Disease D,” is a clear scientific hypothesis that can be taken directly to the lab for validation. This is how AI can truly accelerate drug discovery.
📜Title: Rewarding Explainability in Drug Repurposing with Knowledge Graphs 📜Paper: https://arxiv.org/abs/2509.02276
3. A New Tool for Strained Molecules: strainedSMILES2xyz
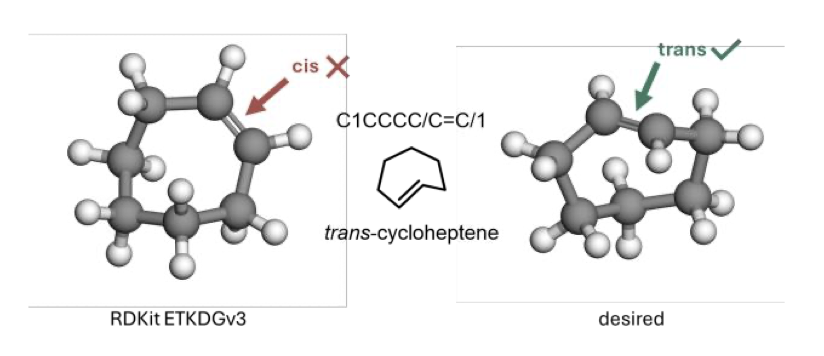
Anyone in computational chemistry has faced this headache: you get a SMILES (Simplified Molecular-Input Line-Entry System) string for a molecule and try to turn it into a reliable 3D structure. But the software gives you a high-energy “monster” with completely distorted bond lengths and angles. This is especially common for molecules with strained rings like cyclobutane or more exotic bridged and spirocyclic systems.
We all know that a bad starting 3D structure means “garbage in, garbage out” for subsequent calculations like molecular docking, dynamics simulations, or anything else that depends on conformation.
What’s the problem?
Common toolkits like RDKit and Open Babel use conformation generation algorithms and force fields trained on large datasets of “normal” molecules. They have a built-in memory for the standard 109.5° bond angle of an sp³ carbon and the 120° of an sp² carbon. But the bonds in strained ring systems are like steel bars that have been bent out of shape; their angles and lengths deviate from the norm. Standard algorithms get confused by this. They either fail with an error or force standard rules onto the molecule, generating a physically impossible conformation.
A Better “Recipe”
The strainedSMILES2xyz workflow from the Svatunek Lab combines existing tools (RDKit, ORCA) in a smarter way. It’s like an experienced chef who knows when to use high heat and when to simmer.
Here’s how it works:
First, loosen RDKit’s constraints. This is the clever part. Instead of abandoning RDKit, it first uses it to generate an initial conformation but with relaxed geometric constraints. This is like telling RDKit, “Don’t be too strict, just give me a rough shape to start with. I know this molecule is a bit weird.”
Second, enumerate the possibilities. A SMILES string can have stereochemical ambiguities. The workflow systematically generates all possible stereoisomers to ensure the correct conformation isn’t missed due to an initial misinterpretation.
Third, run a preliminary optimization with a force field. Once it has the rough 3D structures, it uses a computationally cheap force field (like MMFF94) to quickly screen and optimize them, weeding out the most unreasonable conformations.
Fourth, bring in the heavy machinery: quantum chemistry. This is the key to quality. The best conformation from the previous steps is handed over to the ORCA software package for final geometry optimization using a semi-empirical quantum method (GFN2-xTB). Force fields are based on empirical rules, but quantum chemistry methods calculate from the electronic structure, which gives much higher accuracy for these unusual electronic arrangements and bonding. It’s like using a precision instrument for the final finishing touches on a part to ensure its dimensions are perfect.
How well does it work?
The researchers validated the workflow on a test set of 32 strained and unstrained molecules. The results showed that strainedSMILES2xyz generated the correct geometry in almost all cases, outperforming RDKit, Open Babel, and ChemDoodle 3D. It even handled molecules that caused other tools to fail completely.
The speed is also acceptable, taking only about 20 seconds for the most complex molecules. This means it can be integrated into daily computational workflows, whether for handling a single difficult molecule or converting an entire compound library.
For researchers, having a reliable, automated tool to handle such a fundamental yet critical task saves a lot of time spent manually checking and fixing models. Its value is particularly clear when you’re designing molecules with new, non-classical scaffolds. The tool is released as an open-source Python package and Jupyter notebook, making it easy to use and adapt.
📜Paper: https://doi.org/10.26434/chemrxiv-2025-30dqz 💻Code: https://github.com/Svatunek-Lab/strainedSMILES2xyz