
Table of Contents
- Researchers have developed an AI method called PROTAC-TS, which uses reinforcement learning to design PROTAC linkers, solving a key challenge of cell membrane permeability in drug development.
- Protein language models are not unknowable black boxes; their predictive power comes from an explainable two-step mechanism: identifying local sequence motifs and then activating corresponding structural domains.
- The drGT model not only predicts a drug’s effectiveness on cancer cells but also, like a seasoned researcher, points out the key genes influencing that effectiveness, thus revealing the AI’s decision-making process.
1. AI Designs PROTAC Linkers, Overcoming the Cell Membrane Permeability Hurdle
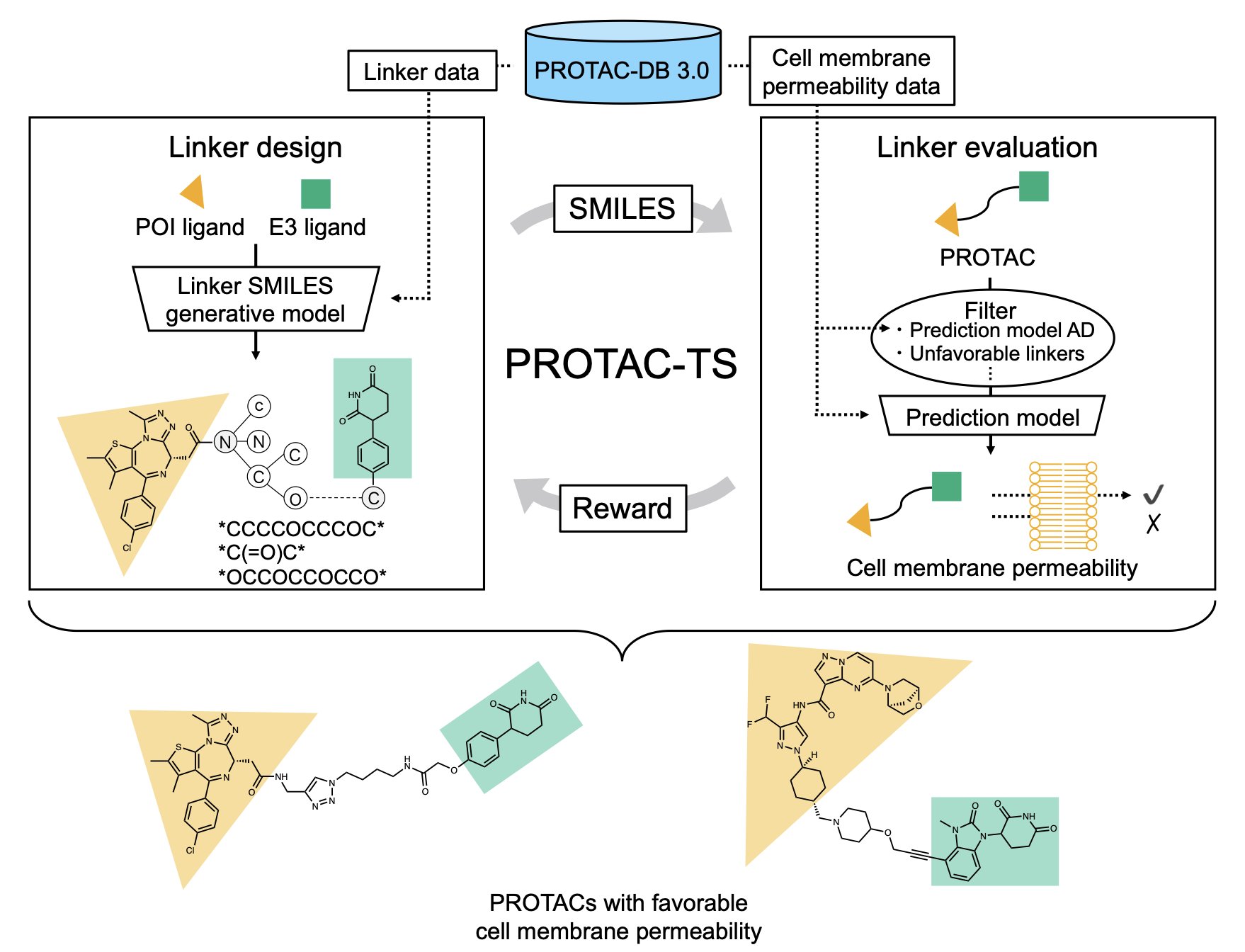
Anyone working with PROTACs (PROteolysis TArgeting Chimeras) knows the core problem: the molecules are too big and often can’t get past the cell membrane. Traditional small-molecule drug design follows Lipinski’s Rule of Five, but PROTACs, with molecular weights often exceeding 800 Da, are firmly “beyond Rule of Five.” As a result, many PROTACs that perform well in cell-based experiments may fail in the body due to poor permeability. The linker is key to tuning the molecule’s physicochemical properties, but designing the perfect one still relies heavily on a chemist’s experience and a lot of trial and error.
This new study proposes an approach called PROTAC-TS to solve this problem with computation.
It works in two steps. First, the researchers built a machine learning model based on the PROTAC-DB 3.0 database specifically to predict PROTAC permeability. The model’s performance was good, with an R² value of 0.710. In drug discovery, for a complex biological property like cell permeability, this is a solid number. It means the model has generally captured the key chemical features that determine PROTAC permeability.
The second step is the core of the method: using Reinforcement Learning to “generate” new linkers. You can think of this as training an AI to play a game of “building blocks.” The AI’s task is to build a linker that is chemically valid and scores high on permeability. Every time it adds a fragment, the permeability prediction model gives it a score. If the score is high, the AI gets a “reward” and will tend to repeat similar actions. After thousands of rounds of self-play and learning, the AI masters the “knack” for designing high-permeability linkers.
But does this method actually work? The researchers performed a critical validation. They had PROTAC-TS try to redesign some known PROTAC molecules with good permeability, such as the clinical-stage molecule KT-474. The results showed that PROTAC-TS successfully generated linkers very similar to these known molecules. It’s like developing a chess AI—if it can reproduce classic moves from game records, it proves it has learned something real. This result shows that PROTAC-TS isn’t just theory; the chemical knowledge it learned aligns with real-world principles.
The method isn’t a silver bullet yet. Its performance is limited by the size of the training dataset, and it currently only optimizes for permeability. A successful PROTAC needs to not only enter cells but also effectively degrade the target protein, have good solubility, and be metabolically stable. The researchers acknowledge that future work needs to integrate these more complex factors.
Still, PROTAC-TS points us in a new direction. It turns linker design from an “art form” into an “engineering task” that can be driven by data. This allows medicinal chemists to focus their energy where it matters most, instead of getting lost in an endless sea of combinations.
📜Title: Data-driven Design of PROTAC Linkers to Improve PROTAC Cell Membrane Permeability 📜Paper: https://doi.org/10.26434/chemrxiv-2025-24kkf
2. Unboxing AI Protein Models: A Two-Step Process to See 3D Structure
We’ve used Protein Language Models (pLMs) to predict structures for years, and they work well. But a question always lingered: how do they actually work? Do they understand the physical and chemical principles of protein folding, or are they just “brute-forcing” it by fitting data? A new study gives us a look at their internal “circuit diagram.”
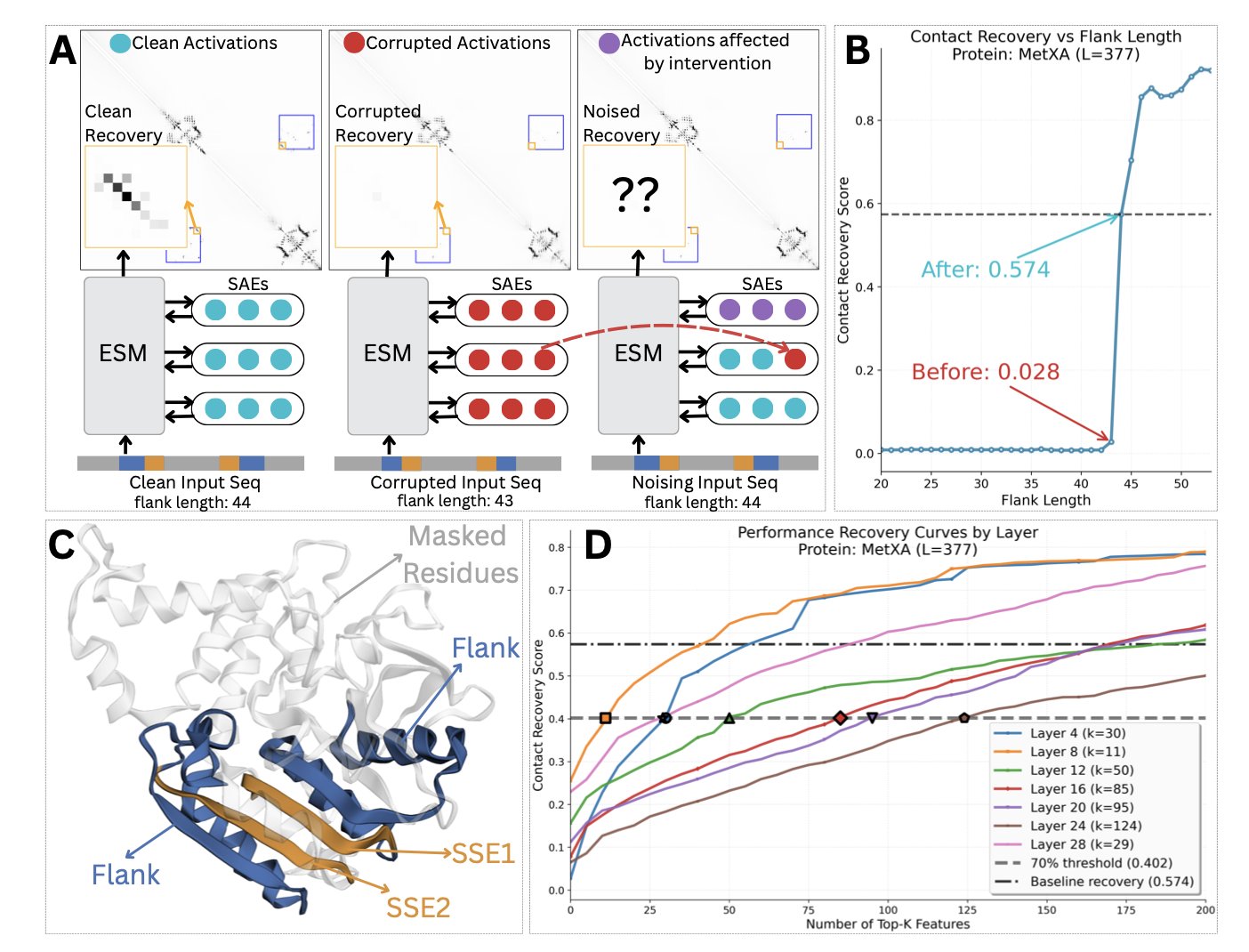
The researchers developed a method to precisely intervene in the model’s “thinking” process. The technique is called causal activation patching in a sparse latent space. It’s like performing precise neurosurgery on the model’s neural network. You can temporarily “turn off” or “activate” a few key neurons and then observe the changes in the model’s predictions. This way, you can find which “neurons” are decisive for the final contact prediction.
They analyzed two proteins, MetXA and TOP2, and found a clear two-step workflow.
First, the early layers of the model act as “motif detectors.” They focus only on short, local amino acid sequence patterns, or “motifs.” These motifs are like special “signposts” along the protein sequence.
Second, once a motif detector is activated, it acts like a switch, “gating” or triggering the middle and later layers of the model. These later layers are “domain detectors,” responsible for recognizing larger protein domains or families.
The whole process is: the model first finds a few key “signposts” in the sequence. Based on the combination of these signposts, it determines which known protein family or domain it belongs to. Once it knows the protein’s overall identity, predicting which amino acids will come into contact with each other becomes much easier.
To validate this finding, the researchers also developed two diagnostic tools. One is the Motif Conservation Test, to confirm that the “motif detectors” are indeed looking for biologically conserved sequences. The other is the Domain Selectivity Framework, to prove that the “domain detectors” have high specificity for particular protein families. The results supported their hypothesis.
This is the first “circuit-level” causal analysis of a protein language model. The model is not a chaotic black box but has evolved an efficient internal mechanism that aligns with biological logic. Understanding this not only helps us build more powerful and reliable predictive models but could also lead to new biological insights.
📜Title: Mechanistic evidence that motif-gated domain recognition drives contact prediction in protein language models 📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.22.671739v1 Code: https://github.com/NainaniJatinZ/plm_circuits
3. drGT: An AI That Not Only Predicts Drug Efficacy but Also Explains Why
Drug development involves massive amounts of data: thousands of compounds on one side, and gene expression profiles and drug sensitivity data for hundreds of cancer cell lines on the other. Connecting this information to predict how a specific drug will affect a specific cancer has always been a core challenge. Many machine learning models can give accurate predictions, but they often act like “black boxes”—we know the result but not the reasoning behind it.
The drGT model offers a new solution.
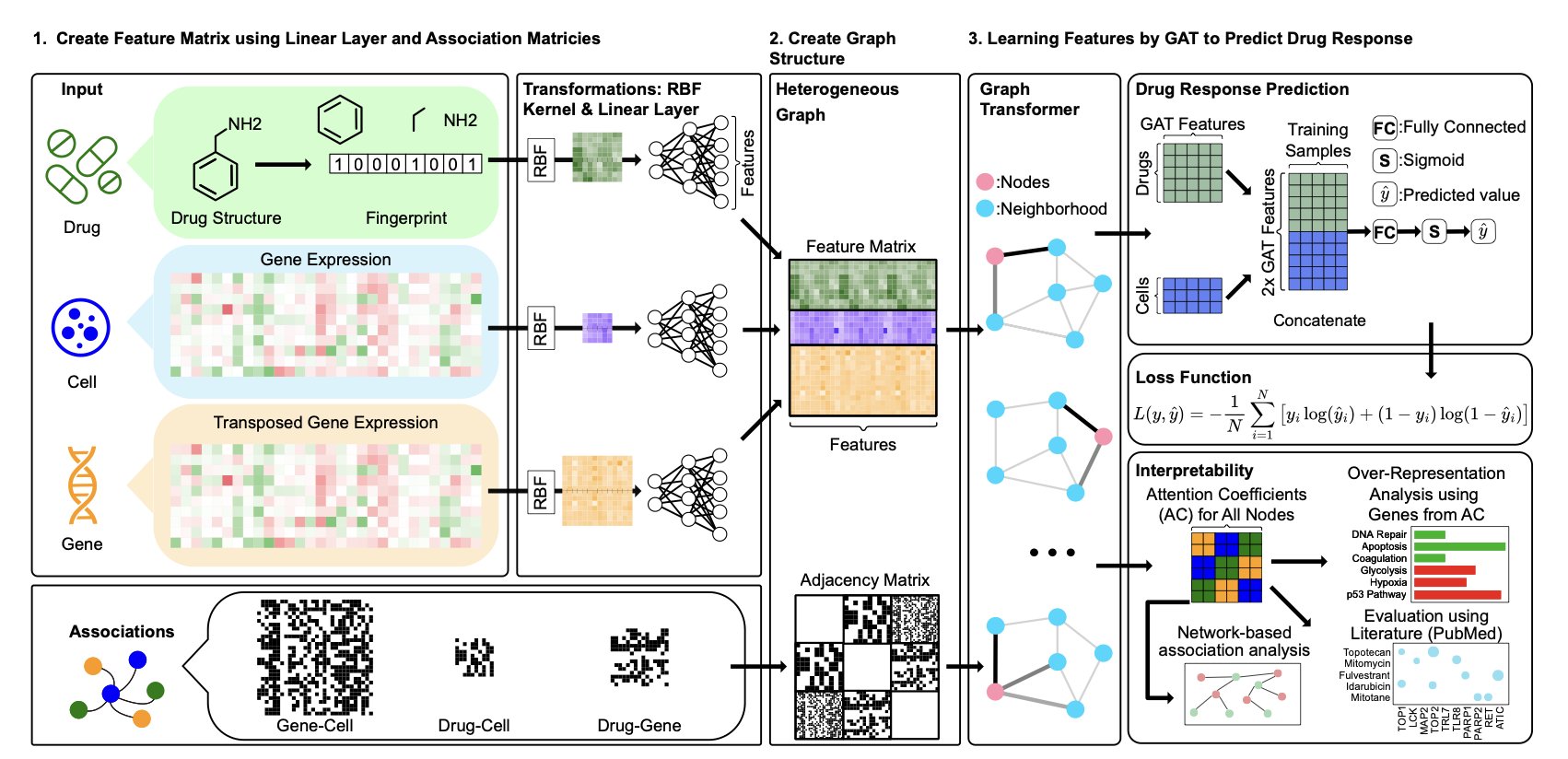
The model’s core is a large Heterogeneous Network. This network is like a complex social graph containing three types of nodes: drugs, genes, and cell lines. The relationships between them are intricate: drugs target specific genes, genes are expressed at different levels in different cell lines, and drugs have varying inhibitory effects on cells. drGT integrates all this information into one large graph.
The technical heart of drGT is a Graph Transformer (GT) architecture. When a drug acts on a cell, it triggers a chain reaction. The Graph Transformer acts like a detective, not treating all clues equally but paying special “attention” to the most critical gene nodes.
This degree of “attention” is quantified by “attention coefficients.” Genes with high attention coefficients are judged by the model to be crucial for the drug response. This reveals the model’s decision-making basis. We get not only a prediction but also a “list of key genes,” pointing to the factors that likely determine the drug’s efficacy.
On standard public datasets like GDSC and NCI60, drGT’s prediction accuracy (AUROC) reached up to 94.5%. When predicting on drugs or cell lines it had never seen before, its AUROC was 84.4% and 70.6% respectively, showing good generalization ability. This capability is essential for exploring new drugs and new indications.
To validate the model’s explainability, the researchers compared the high-attention drug-gene pairs identified by drGT against the PubMed literature database. Over 63% of these associations were either already reported in the literature or could be confirmed by other established drug-target prediction models.
This shows that the associations uncovered by drGT are highly consistent with known biological knowledge, which increases the credibility of the model’s predictions.
A model that can only predict “yes” or “no” has limited use in drug development. But drGT can explain “why” by pointing to potential key genes, which makes it far more valuable. It can guide subsequent experiments and inspire new scientific hypotheses, such as finding new biomarkers or exploring combination therapy strategies. Tools like this that connect computational prediction with experimental validation are exactly what researchers need now.
📜Title: drGT: Attention-Guided Gene Assessment of Drug Response Utilizing a Drug-Cell-Gene Heterogeneous Network 📜Paper: https://arxiv.org/abs/2405.08979v2 💻Code: https://github.com/sciluna/drGT