
Table of Contents
- The new AI model PLMDA-PPI is powerful because it doesn’t just predict if proteins interact. It goes deeper to the physical level, predicting their specific points of contact, making it unusually robust when facing new proteins.
- Researchers use pretrained deep learning models to intelligently select initial drug combinations and doses, solving the “cold-start” problem of personalized drug screening when no prior data exists.
- The AlphaPPIMI framework improves the accuracy and generalization of PPI modulator prediction by combining multimodal pretrained models and an adversarial network, providing a new tool for targeting protein interfaces.
- MolReasoner uses a two-step “lecture and tutor” method to force AI to stop memorizing and start thinking like a chemist. This makes its answers not only more accurate but, for the first time, explains “why.”
- HyperLab aims to package complex computational workflows into a web tool for medicinal chemists, balancing speed and accuracy.
1. AI Predicts PPIs: Not Just “Yes/No,” but Seeing the “Handshake”
In computational biology, we’re used to prediction models. Most of them act like students who only cram for exams. They perform well on the training set and similar data, but fall apart when they encounter something new, like a protein with an unfamiliar sequence. This is especially true for predicting protein-protein interactions (PPIs), and it’s a real problem.
Now, a new paper introduces the PLMDA-PPI model, which seems like the student who actually understands the physics.
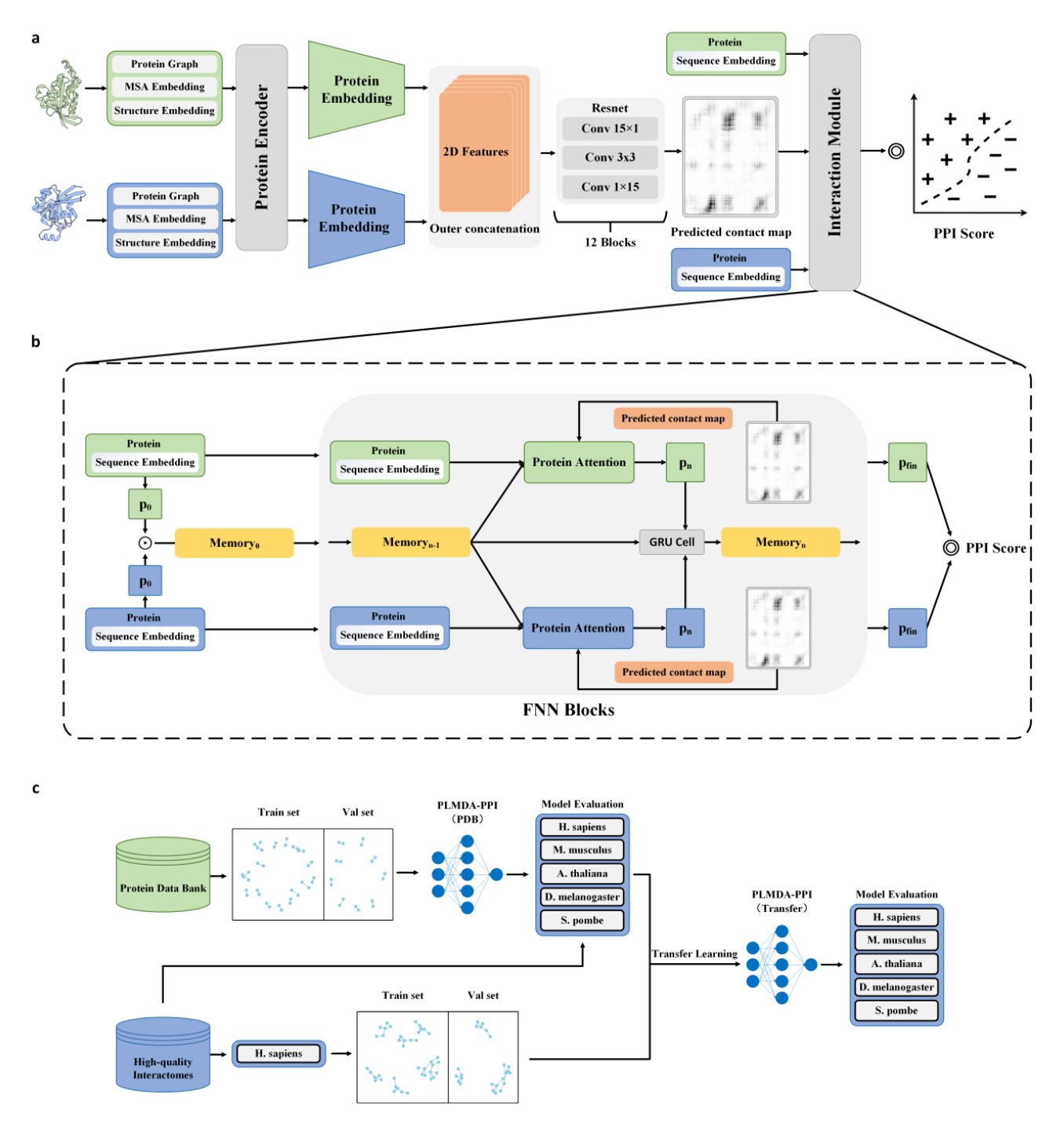
The model’s strength is its approach. Instead of brute-force fitting, the researchers designed a “mechanism-aware” framework. It doesn’t just answer “Will these two proteins bind?” (a Yes/No question). It must also identify which specific amino acid residues form the “handshake.”
It’s like not only guessing if two people are friends but also describing the details of their last meeting. If you can get the details right, your guess is much more likely to be correct. This dual task forces the model to understand the physical and chemical nature of protein interactions, rather than just memorizing that certain sequence fragments often appear together.
To do this, the researchers used a solid method: they trained the model on protein complexes from the PDB database with experimentally verified 3D structures. This is gold-standard data. The model learns not just abstract sequence relationships but also real geometric and physical contacts.
The results are a clear step up. On tough test sets designed to challenge generalization (using proteins with very low sequence similarity), older state-of-the-art models failed. But PLMDA-PPI held strong, showing much higher accuracy and robustness. This proves it learned the underlying principles.
For drug developers, this is very valuable. PPIs are a rich source of “undruggable” targets, as many diseases involve abnormal protein interactions. But their binding interfaces are often large and flat, making them difficult for small molecules to target. A tool that can precisely predict binding “hotspot” residues is like a high-resolution treasure map for drug designers. We can focus our efforts on designing molecules—whether small molecules, peptides, or PROTACs—that fit perfectly into these key contact points.
So, PLMDA-PPI isn’t just another PPI predictor. It offers a new way of thinking about modeling—teaching AI to think like a structural biologist. This approach could be extended to predict interactions between other biological macromolecules, like RNA and DNA. That’s what’s truly exciting.
📜Title: Mechanism-Aware Protein-Protein Interaction Prediction via Contact-Guided Dual Attention on Protein Language Models
📜Paper: https://www.biorxiv.org/content/10.1101/2025.07.04.663157v2
💻Code: https://github.com/ChengfeiYan/PLMDA-PPI
2. AI Guides Personalized Drug Combinations, Solving the “Cold-Start” Problem
In personalized cancer therapy, we often face a tough question: for a new patient, with hundreds of anticancer drugs available, which combinations should we test first? It’s like trying to find treasure in a new city without a map. You can’t search every street; time and resources are limited. This is the “cold-start” problem in drug screening.
The authors of this paper wanted to solve this navigation problem. They trained a deep learning model that acts as a pre-drawn “drug-efficacy map.”
First, they trained the model on massive amounts of historical data, including the effects of various drug combinations at different doses on tumor cells. By learning from this data, the model generates a unique mathematical representation, or “embedding,” for each drug combination. You can think of this embedding as a coordinate on the map. Drug combinations with similar mechanisms of action will have coordinates that are close to each other.
With this map, we can plan our “exploration route” for a new patient. We can’t test every point on the map, so we need to select a few representative ones. The researchers used a clustering algorithm called K-medoids. This algorithm automatically identifies several core regions on the map and picks the most representative point from each. This is like planning a trip where instead of visiting every corner of a city, you select a few key spots like the business district, the old town, and the art district. This ensures our initial tests cover diverse mechanisms of action, greatly reducing the risk of betting on the wrong drugs.
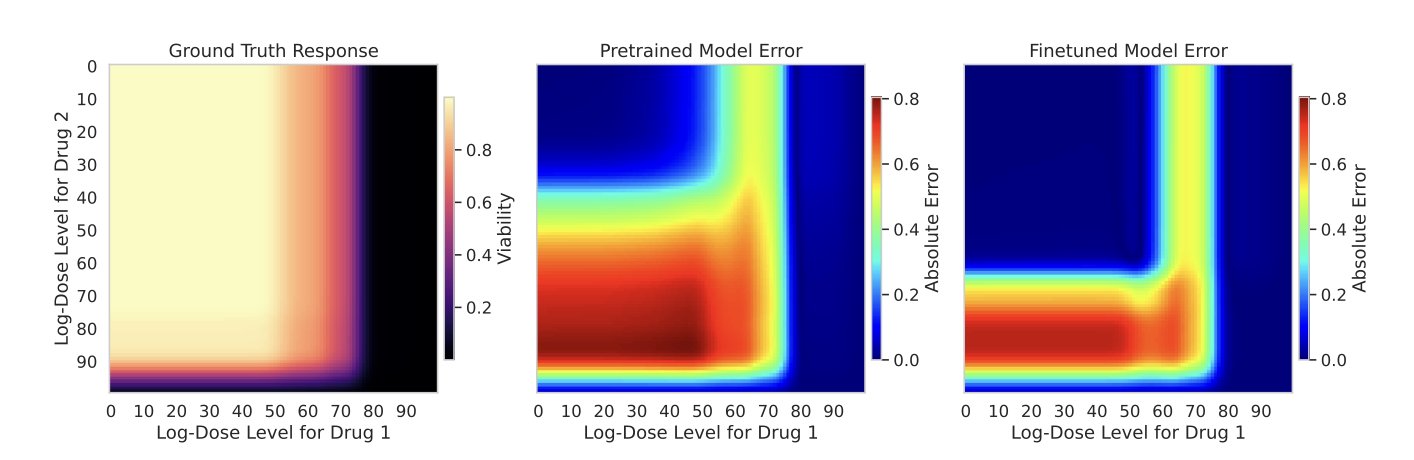
Once the combinations are chosen, the next step is to decide which doses to test. The traditional approach might involve testing a wide dose range, which is also resource-intensive. Here, the model helps again. It not only predicts whether a combination will be effective but also predicts the entire dose-response curve.
Anyone in drug development knows that not all points on this curve are equally informative. The area with the steepest slope, usually around the IC50 value, contains the most information. By analyzing the shape of the predicted curve, the model assigns an “importance score” to each dose point. A higher score means the point is more critical for determining the drug’s potency. This allows us to focus our limited experimental resources on these high-value dose points, getting the most information about drug sensitivity from the fewest experiments.
To validate this strategy, the researchers ran retrospective simulations using large drug screening databases. They pretended they had no information about the samples and used their method to select initial experiments. The results showed that compared to baseline methods like random selection, this new strategy found effective drug combinations faster and more accurately.
This is a practical advance for personalized medicine. It means we can screen potential combination therapies for patients more efficiently in the early stages of clinical research and make decisions faster.
3. AI Predicts PPI Inhibitors: A Deep Dive into the AlphaPPIMI Paradigm
Targeting protein-protein interactions (PPIs) in drug discovery has always been a tough challenge. These interaction surfaces are often large and flat, unlike the well-defined active pockets of traditional enzymes, making it very hard to find small molecules that can block them effectively. So whenever a new computational tool claims a breakthrough in this area, we listen closely.
A recent paper from Liu and his team on AlphaPPIMI is exactly that kind of progress. They built a new deep learning framework with a clear goal: to predict which small molecules can modulate PPIs more accurately and reliably.
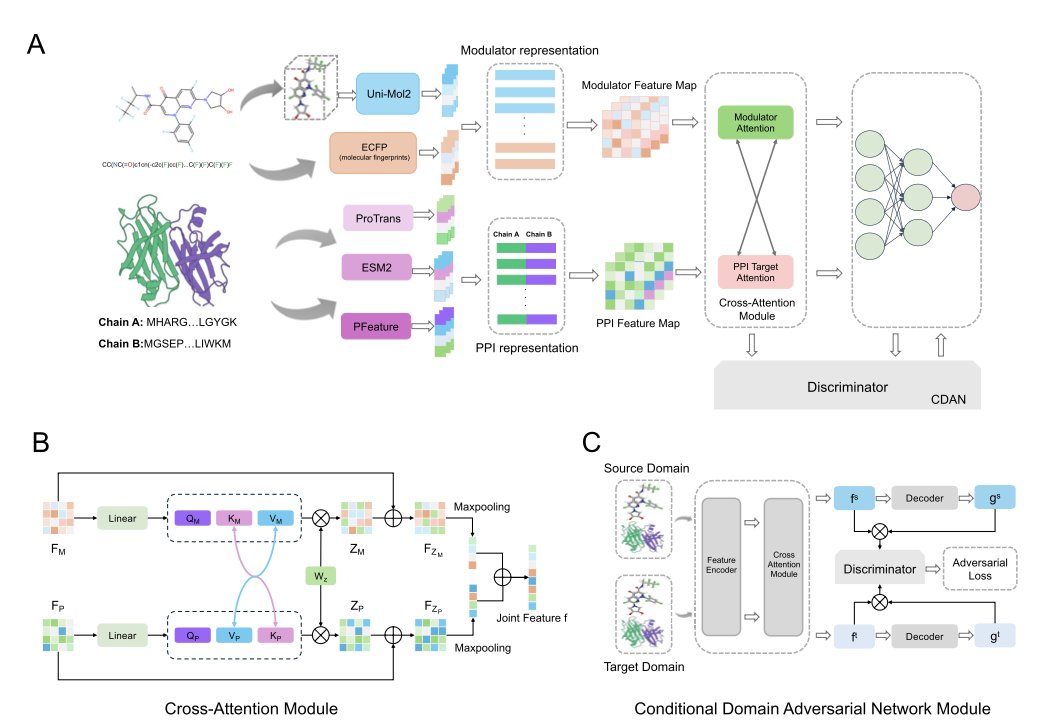
The model’s cleverest part is its core—a custom cross-attention module. Traditional models often look at the small molecule and the protein separately, then simply combine the information. AlphaPPIMI works differently. Imagine it as a translator fluent in both “chemistry” and “biology.” It first prepares the “native language” materials: 3D structural information for the small molecule (from the Uni-Mol2 model) and sequence information for the protein (from the ESM2 and ProTrans models). Then, the cross-attention module gets to work. It lets the features of the small molecule and the protein interface “talk” to each other, dynamically analyzing how a specific group on the molecule affects an amino acid at the protein interface, and vice versa. This way, the model establishes a deep, context-aware interaction model instead of looking at the problem in isolation.
Anyone in computer-aided drug discovery (CADD) knows that model generalization is a big issue. A model might perform well on its training data, but if it fails on a new, unseen target family, its practical value is limited. The researchers addressed this using a technique called Conditional Domain Adversarial Networks (CDAN). The idea is interesting. It adds a “troublemaker” network alongside the main model. The main model’s job is to predict the interaction between the molecule and the target. The troublemaker’s job is to guess which protein family the interaction data came from. The main model’s goal then becomes twofold: make accurate predictions while also “tricking” the troublemaker so it can’t guess. If it succeeds, it means the features the main model learned are universal binding principles, not just rules specific to one protein family. This greatly enhances the model’s reliability when facing new targets in the real world.
Of course, theory needs to be backed by results. The authors tested AlphaPPIMI extensively on their own benchmark dataset, where it outperformed existing methods. More importantly, they conducted case studies. For instance, they used the model to screen for inhibitors of the Hsp90-Cdc37 complex, a known valuable but difficult PPI target. The model successfully identified several promising candidate compounds. They also applied the method to the gp120/CD4 interaction, which is directly relevant to HIV drug development. These examples show that AlphaPPIMI is not just an academic model. It has the potential to help us prioritize the most promising molecules for synthesis and testing from virtual libraries of millions, which can save real time and money in R&D.
📜Title: Alphappimi: a comprehensive deep learning framework for predicting PPI-modulator interactions
📜Paper: https://doi.org/10.1186/s13321-025-01077-2
4. AI Stops Memorizing Answers: MolReasoner Teaches It Chemical Reasoning
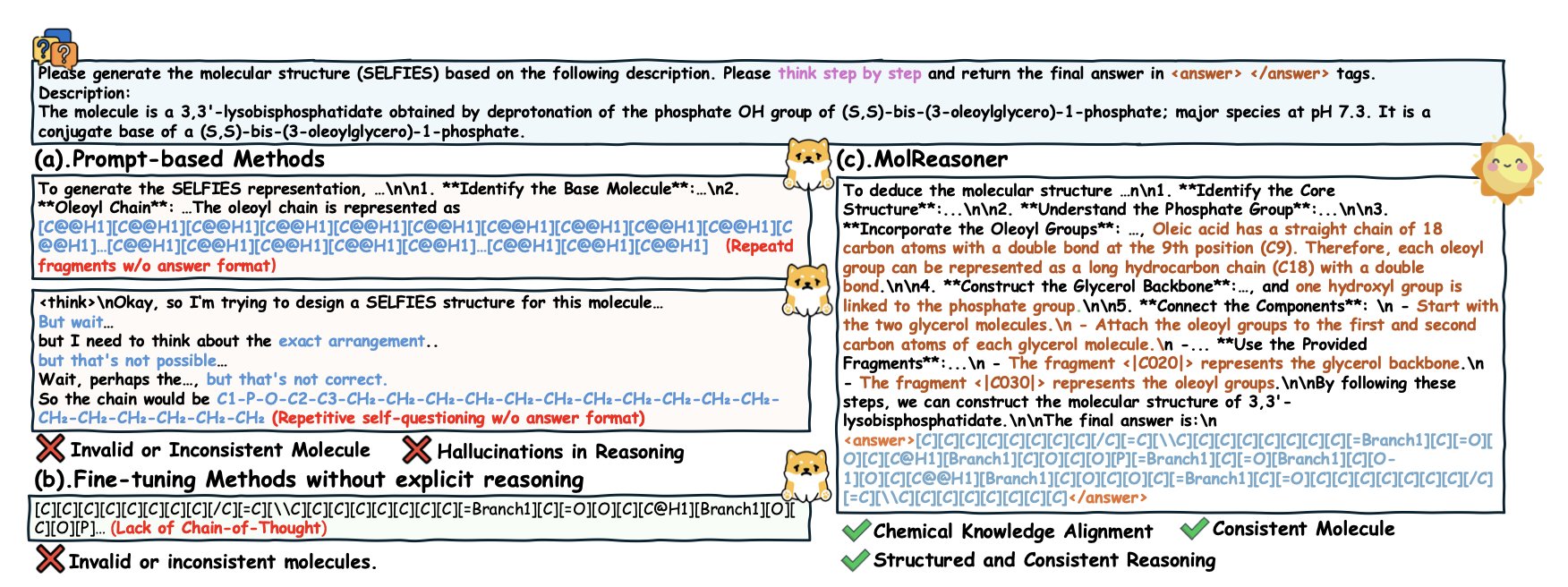
The large language models we have today are like students with excellent memory but little understanding. Ask one about a molecule’s properties, and it can pull a decent answer from its vast database. But ask it “why,” or ask it to design a new molecule based on chemical principles, and it often reveals itself as a machine that memorizes answers, not a chemist that solves problems.
MolReasoner’s strategy is much like how we train a good student: first attend class, then do homework, and finally get a good tutor.
Phase 1: The Lecture (Mol-SFT)
The goal here is to first teach the AI the “format” for solving problems and basic “chemical grammar.” The researchers didn’t just throw messy real-world data at the model. Instead, they had a “PhD student” like GPT-4o generate a large set of problem-solving steps with a “Chain-of-Thought.” For example, when describing a molecule, it might first state, “This molecule contains an electron-donating methoxy group and an electron-withdrawing nitro group…” before giving its conclusion.
But they didn’t fully trust this PhD student. They also had “chemistry experts” (either humans or other validation tools) grade this work to ensure every reasoning step was chemically sound. This created a high-quality “textbook” full of correct examples. Using this textbook for supervised fine-tuning (SFT), the AI first learned to speak like a person, or more specifically, like a chemist.
Phase 2: One-on-One Tutoring (Mol-RL)
Knowing the format isn’t enough; you have to be fast and accurate. That’s where the second phase, reinforcement learning (RL), comes in. This is like a strict tutor watching the AI solve problems. The “reward function” it uses is clever. It doesn’t just check if the final answer is right; it also examines each step of the AI’s reasoning. If the AI’s logic is clear and follows chemical principles, it gets a high reward. If it starts making things up, it gets no reward, even if it guesses the right answer in the end.
This “process and outcome” approach to tutoring forces the AI to learn real chemical logic instead of taking shortcuts.
MolReasoner showed a dominant advantage in two tasks: molecular captioning (writing a chemical description for a molecule) and text-based molecular generation (drawing a molecule from a description). Its generated descriptions are more accurate, and the molecules it designs are more reasonable and effective.
📜Title: MolReasoner: Toward Effective and Interpretable Reasoning for Molecular LLM
📜Paper: https://arxiv.org/abs/2508.02066v1
💻Code: https://github.com/545487677/MolReasoner
5. HyperLab Review: An AI Drug Platform with Accuracy Approaching AF3
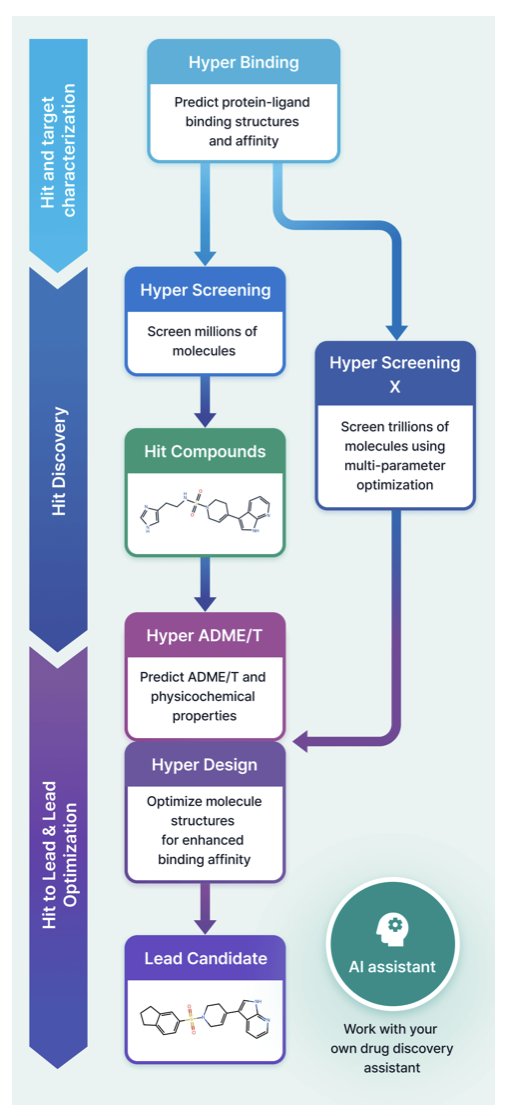
There’s no shortage of AI drug discovery platforms; new tools seem to appear every week. So what makes HyperLab, developed by the HITS team, special? Its positioning is clear: to be a simple, web-based tool that integrates the entire workflow from hit screening to lead optimization. The target users are frontline R&D scientists, especially medicinal chemists and biologists who aren’t experts in computational chemistry.
Let’s start with its core feature: Hyper Binding. This module predicts how a small molecule binds to a protein target. This is the starting point for all structure-based drug design (SBDD), and its accuracy directly determines whether subsequent optimization efforts are headed in the right direction. The researchers tested it using the public benchmark PoseBuster v2, and the results showed an accuracy of 77%.
This is an interesting number. Traditional molecular docking software like Glide or GOLD typically performs in the 60-70% range on this dataset. One of the top models today, AlphaFold3, has an accuracy of 84%. HyperLab’s 77% sits right in the middle—better than traditional methods, and not far behind the top model. But its advantage is speed. For a medicinal chemist, this means you can test dozens of newly designed molecular structures in a morning, instead of sending one molecule to the computational team and waiting a day. Trading a bit of accuracy for a huge gain in speed is a good deal in the early exploration phase.
Another highlight of the platform is Hyper Screening X. It offers a virtual library of 11 trillion molecules. What does that number mean? Traditional physical compound libraries, like Enamine REAL Space, are in the tens of billions, which is already massive. Eleven trillion represents a vast expansion of chemical space. And this isn’t just a static library. It uses generative AI to optimize within this huge chemical space, helping you find entirely new and active molecular scaffolds. This is very attractive for anyone searching for first-in-class drugs.
HyperLab also integrates molecular design (Hyper Design) and ADME/T prediction (Hyper ADME/T). This creates a closed loop. You can find a hit compound with Hyper Screening, analyze its interaction with the target using Hyper Binding, modify its chemical structure in Hyper Design, and then immediately predict properties like solubility and metabolic stability with Hyper ADME/T. The entire workflow can be done in a single web interface, without switching between different software.
Of course, claims need proof. The researchers presented an internal case study where they used the HyperLab platform from scratch and ended up with nanomolar-level active compounds, all verified by experiments. While this is internal data and should be viewed with caution, it at least demonstrates that the workflow is viable in practice.
Finally, the platform includes a built-in AI assistant. It can help you search databases, create plots for analysis, or provide guidance when you’re unsure how to use a feature. This further lowers the barrier to entry.
Overall, HyperLab’s approach isn’t about pushing a single metric to its limit. Instead, it strikes a good balance between accuracy, speed, and ease of use. It aims to turn computer-aided drug discovery from a specialized skill into a tool that every drug developer can use.
📜Title: Accelerating Drug Discovery with HyperLab: An Easy-to-Use AI-Driven Platform
📜Paper: https://www.biorxiv.org/content/10.1101/2025.08.31.672525v1