
Table of Contents
- OwkinZero used reinforcement learning on a high-quality, verifiable Q&A dataset to prove that small, specialized AI models can beat larger, more general commercial models at biological reasoning.
- The generative AI model HemePLM-Diffuse is 100 times faster than traditional molecular dynamics. Instead of simulating frame by frame, it directly generates the entire dynamic process of a protein-ligand interaction.
- Don’t get hung up on complex models. The real win comes from a hybrid strategy: use deep learning for ADME and classical methods for potency. But most importantly, fix the problems in your data first.
1. AI for Biological Reasoning: Small Models Beat Big Models
Asking a Large Language Model (LLM) a serious biology question can be a strange experience. It feels like talking to a brilliant intern who has read everything. It cites papers and sounds confident, but you can’t shake the feeling it doesn’t really understand. It’s mimicking massive amounts of text, not reasoning from first principles.
This is the LLM’s “biological reasoning blind spot.” And a new paper hits it right on the mark.
Stop Rewarding Fancy Explanations
In the past, AI training often rewarded the “chain of thought.” It was like a teacher grading homework: even if the final answer was wrong, you got partial credit for showing your work.
The researchers behind OwkinZero took a stricter, more practical approach: only reward correct answers.
They used a strategy called Reinforcement Learning from Verifiable Reward (RLVR). Think of it as an endless multiple-choice test. For every question the AI answers, the system checks it against a reliable external knowledge source. Correct answers get a reward; wrong ones get a penalty.
This direct method forces the AI to focus all its energy on getting the right conclusion, not on crafting a convincing explanation.
The Real Secret Weapon: A Perfect Textbook
A good exam needs good study materials, and this is where the work really shines.
Instead of scraping data from the internet, the team brought in experts to build eight benchmark datasets with over 300,000 high-quality question-and-answer pairs. These are real problems that researchers face: What’s the druggability of a target? Should we use a small molecule or an antibody? What cellular response will a drug cause?
They essentially wrote the most authoritative textbook and practice exam for the AI in biomedical science.
The Results Were Surprising
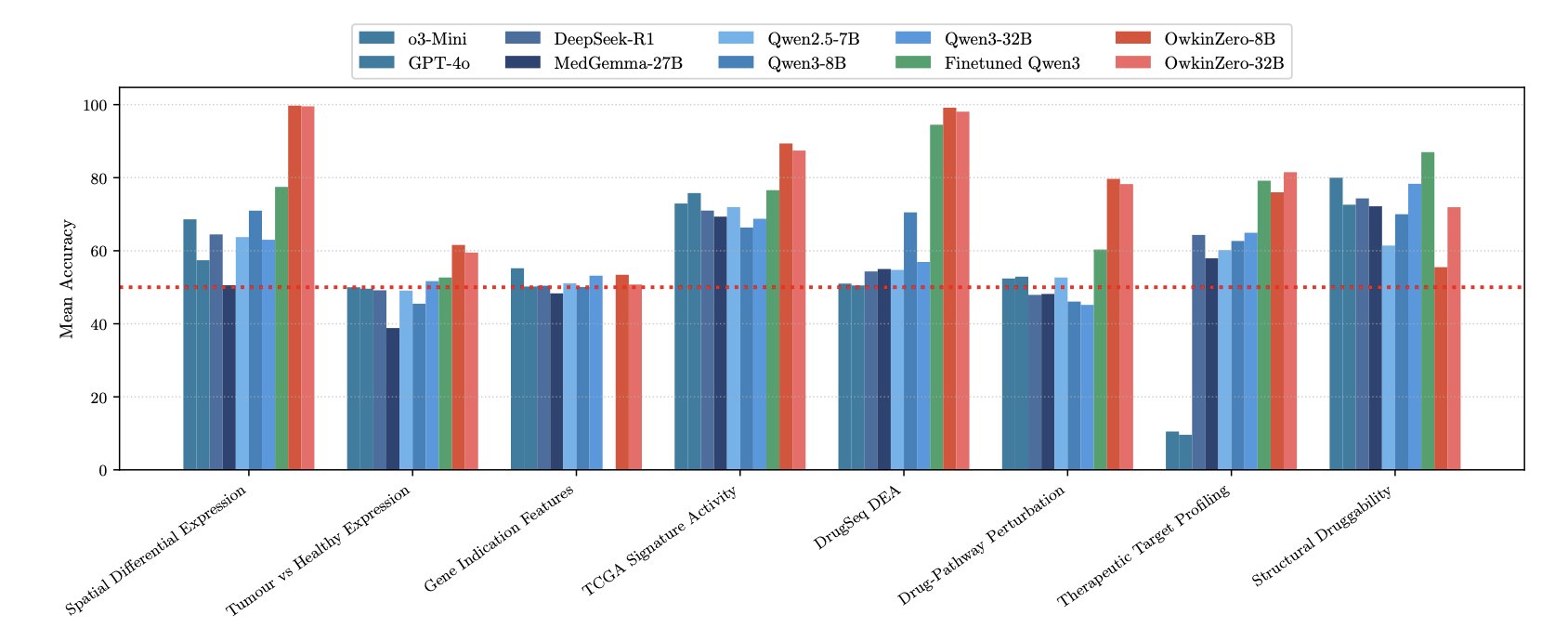
So how did OwkinZero, a medium-sized model trained with this method and material, perform?
It consistently beat general-purpose commercial LLMs that are far larger and more expensive to run on biological reasoning tasks.
It’s like a specialized athlete, with expert coaching, beating an all-around star who is good at everything but a master of none.
Even more interesting was the “generalization effect.”
The researchers found that an expert model trained only on “target druggability” questions got better at its specialty, but it also improved at a task it had never seen before: predicting “drug perturbation effects.”
This is counterintuitive. It’s like a medical student who only studies cardiology but also gets better grades in nephrology. It suggests the model learned underlying principles of physiology and pathology that apply across medicine, not just isolated facts about the heart.
OwkinZero seems to be starting to truly “think” about biology.
Maybe we don’t need to wait for an all-knowing GPT-6. With focused, high-quality data and the right training methods, we can build expert AI systems today that are more powerful and reliable than general models for specific scientific domains.
📜Title: OwkinZero: Accelerating Biological Discovery with AI 📜Paper: https://arxiv.org/abs/2508.16315
2. AI Directs a Molecular Movie: 100x Faster Than MD
Anyone who has run a Molecular Dynamics (MD) simulation knows the feeling. MD is the only “camera” we have to see how a drug molecule squeezes into its target. But this camera shoots in slow motion and has a short battery life.
Drug binding or unbinding can take microseconds or even milliseconds. But a supercomputer cluster running for months might only capture a few hundred nanoseconds. We have a tool with atomic resolution, but we can’t film the whole movie.
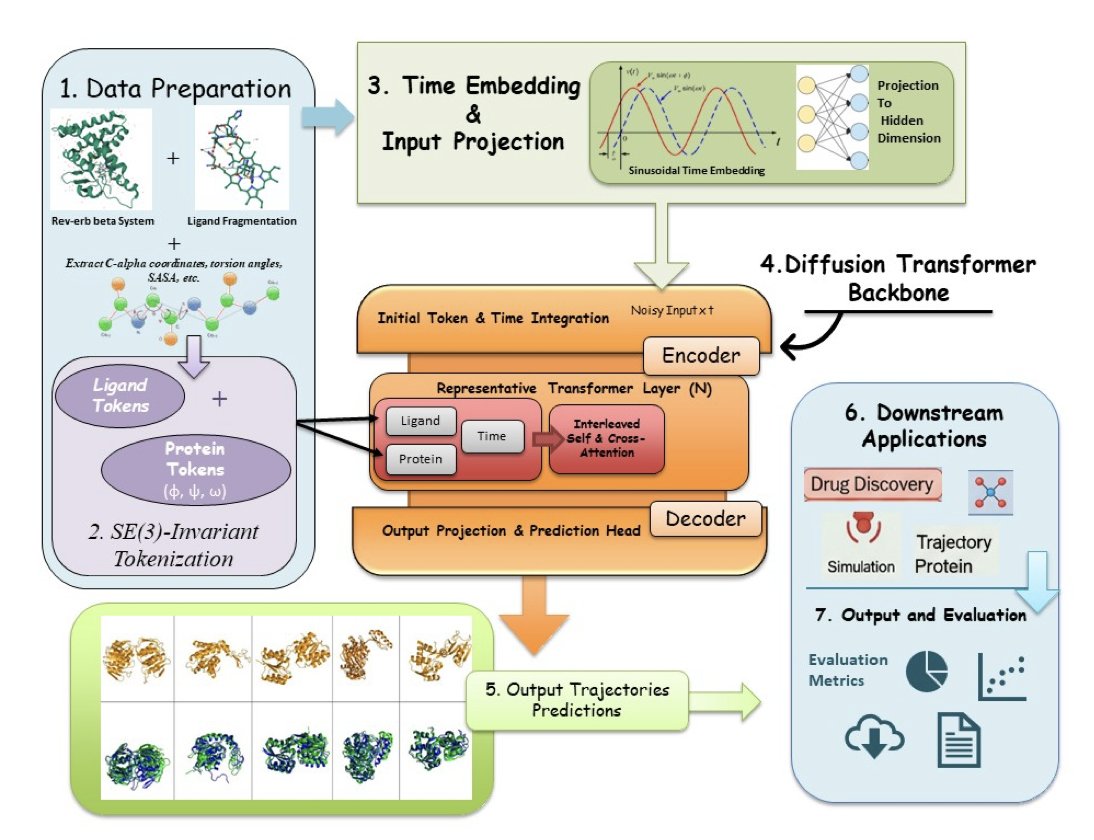
HemePLM-Diffuse chooses to be the “director” instead of the “camera operator.”
How AI Learned to Direct
A great director doesn’t need to operate the camera. They’ve watched countless films and understand the internal logic of storytelling.
HemePLM-Diffuse, a generative Transformer model, works the same way. It doesn’t calculate physics directly. Instead, it learned from vast amounts of MD simulation data, mastering the patterns of protein-ligand interactions.
It knows which chemical groups tend to interact with which amino acid residues. It knows how flexible loops usually respond when a ligand gets close.
So, it no longer needs to shoot frame by frame. Just give it a “script”—like the starting and ending states of protein A binding with ligand B—and the AI can generate the complete, high-probability dynamic process connecting them.
The process is closer to how DALL-E generates an image than a physical simulation. Its speed comes from skipping the tedious, step-by-step calculations.
How Good Is the “Director”?
To test its reliability, researchers benchmarked it on several key tasks:
Ligand Inpainting: They removed the ligand from a trajectory and asked the model to put it back. HemePLM-Diffuse achieved an average RMSD of just 0.91 Å, outperforming previous models and showing it understands the relationship between the ligand and the protein.
Trajectory Upsampling: Given only the first and last frames, the model had to fill in the middle. It also performed well here, with an average per-frame RMSD of 1.03 Å. This is valuable for simulating expensive systems.
Transition Path Sampling: It generated transition paths from state A to state B. Its TPS score was 0.95, indicating the generated paths are physically plausible.
In terms of speed, simulating 1 nanosecond of dynamics for a large system containing heme took just 12 minutes. A traditional MD method would take days. This is a speedup of over 100 times, which is a qualitative shift.
The current version does not explicitly account for the effects of solvent (water molecules), which is a simplification for many systems.
HemePLM-Diffuse opens a new path. It brings us one step closer to the goal of quickly and broadly predicting drug kinetics (not just thermodynamics) in the early stages of drug design.
📜Title: HemePLM–Diffuse: A Scalable Generative Framework for Protein–Ligand Dynamics in Large Biomolecular System 📜Paper: https://arxiv.org/abs/2508.16587v1
3. AI in Pharma: Old Methods Aren’t Dead, Deep Learning Only Wins on ADME
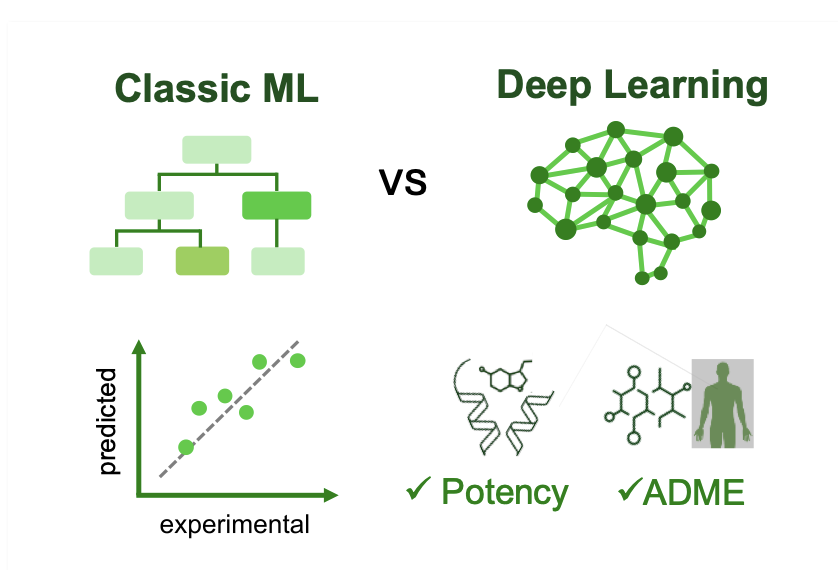
A duel is underway in computational drug discovery. In one corner are the time-tested classical machine learning methods, like random forests and gradient boosting machines. In the other is deep learning, with its impressive array of complex neural networks.
A paper from the “Polaris Antiviral Challenge” provides a detailed referee’s report on this duel. The conclusion might surprise deep learning’s supporters.
Predicting Potency: The Old Guard Still Has It
In the core task of predicting compound potency, the result was a draw.
Classical machine learning methods performed just as well as deep learning models that require immense computational power. This suggests that for tasks like predicting how strongly a molecule binds to a target, more model complexity does not equal better performance.
Predicting ADME: Deep Learning’s Home Turf
However, in the more complex domain of predicting pharmacokinetics—Absorption, Distribution, Metabolism, and Excretion (ADME)—deep learning scored a decisive victory.
ADME involves many factors and non-linear problems. A molecule’s solubility, permeability, and metabolic stability are determined by a combination of many subtle physicochemical properties. Identifying these complex patterns is exactly where deep learning excels.
The Real “Secret to Winning”
The study found that data processing methods had a bigger impact on the results than the choice of model.
The key was handling “activity cliffs.” An activity cliff is when two molecules with nearly identical chemical structures have vastly different biological activities. This kind of data can seriously confuse a model.
The researchers used a simple strategy: before training, they identified these confusing data pairs and temporarily masked them. This data preprocessing step significantly improved the prediction accuracy of both classical and deep learning models.
Instead of arguing about which algorithm is superior, we should spend more time cleaning up the problems in our data.
Another finding was that learned embeddings generated by deep learning models generally performed worse in this challenge than the long-established classical chemical descriptors. This is a reminder that new methods are not always better than tools that have stood the test of time.
In summary, this duel has no absolute winner. The best strategy is a practical combination of both: use deep learning for complex ADME predictions, while sticking with the robust classical methods for potency prediction. But no matter which model you choose, the first job is to inspect and clean your data.
📜Title: Deep Learning vs Classical Methods in Potency & ADME Prediction: Insights from the Polaris Antiviral Challenge 📜Paper: https://doi.org/10.26434/chemrxiv-2025-64fcb