
Table of Contents
- PARSE focuses on conserved local microenvironments in proteins, not their overall structure, to more accurately predict function and active sites. It’s especially good at studying the “dark proteome.”
- The Honeybee framework integrates various types of cancer data. The research found that simple clinical text has more predictive power than complex genomic or imaging data.
- Boltzina uses a fast but rough tool for initial work, followed by a slow but precise AI for the final evaluation, balancing speed and accuracy in virtual drug screening.
1. AI Predicts Protein Function: Look Locally, Not Globally
AlphaFold has mapped the structures of hundreds of millions of proteins, giving us a map of the universe. But the functions of most “planets” (proteins) on this map are still unknown. They make up the vast “dark proteome.” We have their 3D structures, but we don’t know what they do.
In the past, we predicted protein function mainly by comparing amino acid sequences with BLAST or overall 3D structures with Foldseek. Both tools rely on overall similarity. If two proteins have very different sequences or overall folds, they’re considered unrelated. This is like trying to determine family ties by facial resemblance, which might miss distant relatives who look different but share a common family trait.
A paper in PNAS suggests that AI needs a magnifying glass to spot these local “family traits.”
Identifying Functional “Fingerprints”
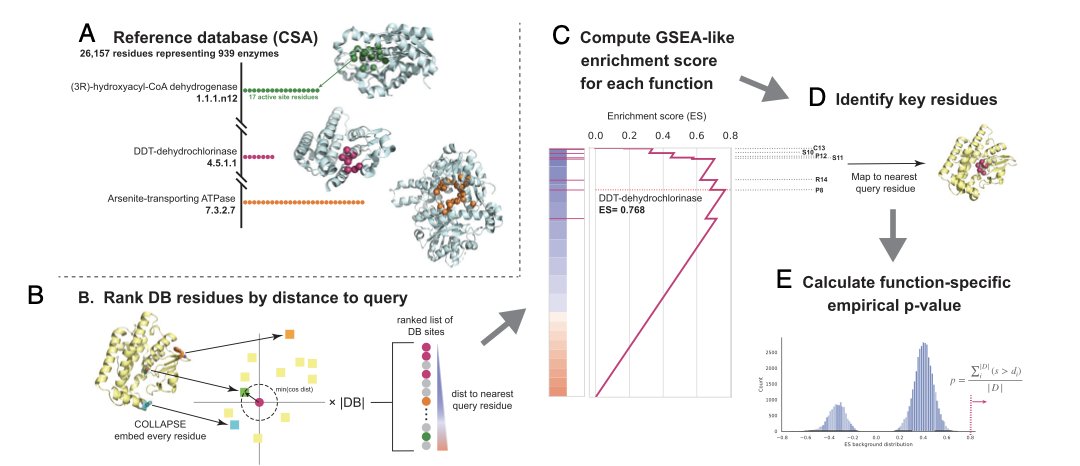
The new method, PARSE, is based on a biological intuition: a protein’s function is determined by its small, evolutionarily conserved active or functional sites, not its large, variable overall structure.
This site is the protein’s “fingerprint.” PARSE’s workflow skips comparing the whole structure:
First, it breaks down a protein into countless overlapping local microenvironments, each centered on a single amino acid.
Next, it uses a tool called COLLAPSE to convert the 3D geometric information of each local environment into a digital “fingerprint” (an embedding).
Finally, it directly compares the local “fingerprints” of different proteins in a large database, looking for highly similar regions.
Pinpointing Functional Sites
By identifying these “fingerprints,” PARSE’s predictive power is much improved. Its performance in predicting overall protein function is on par with complex deep learning models, with an F1 score over 85%.
PARSE’s strength is its precision. While predicting that “a protein is a kinase,” it can also identify the specific amino acids that form its active site with high confidence. Its localization accuracy surpasses existing methods like DeepFRI.
This kind of precise target information is critical for drug development. Researchers need an exact “target map” to guide site-directed mutagenesis or drug design, not a vague functional label.
The method is fast enough to scan the entire human proteome. The researchers used this to find new potential functional sites in some proteins with unknown functions.
This work is an example of how to build AI that better reflects biological principles. It shows that studying key local features can sometimes be more effective than analyzing the overall structure.
📜Title: Protein functional site annotation using local structure embeddings 📜Paper: https://www.pnas.org/doi/10.1073/pnas.2513219122
2. AI in Cancer Research: Clinical Data Is King
The field of cancer research has a huge amount of data, but unlocking its value is a challenge. A single cancer patient generates multiple types of data: clinical notes from doctors, Whole Slide Imaging (WSI) from pathology, CT or MRI scans from radiology, and genomic sequencing maps. This data is scattered across different departments, like experts who don’t speak the same language, making it hard to get a complete picture of the patient.
The industry has been hoping for a “master translator” to integrate all this information and provide a unified view of the patient. The Honeybee framework is an attempt to do just that.
How Honeybee Assembles the Full Puzzle
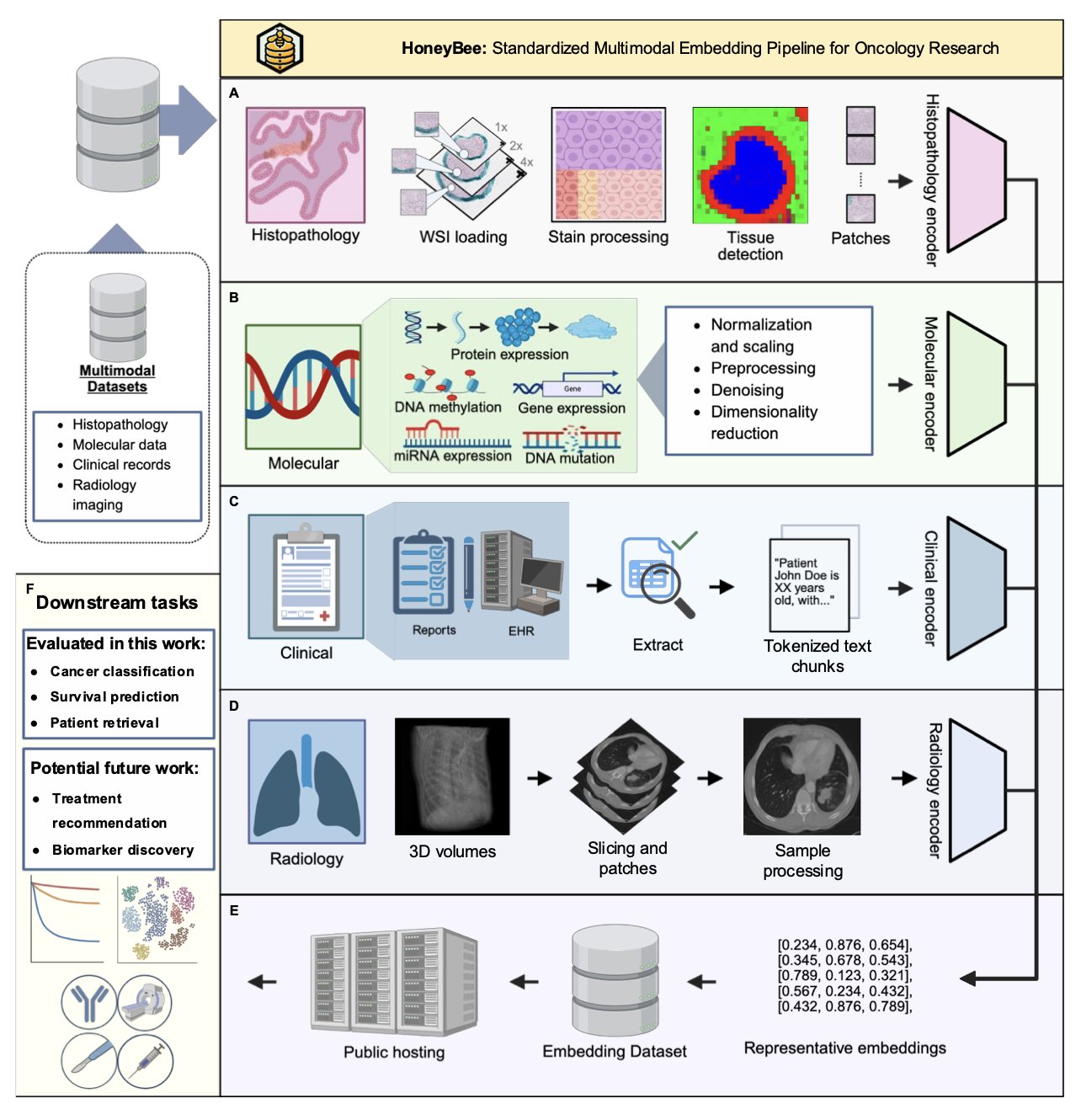
Honeybee is an open-source framework that can process clinical text, pathology images, radiology images, and molecular data. Using a series of AI foundation models, it generates a single “digital fingerprint” (embedding) for each patient.
This “digital fingerprint” can be used for many things, like predicting survival time, classifying diseases, or quickly finding similar cases in a database.
Intuitively, integrating all data should lead to better results. But after testing it rigorously on data from over 10,000 TCGA patients, the researchers made a surprising discovery.
The Strongest Predictor: Clinical Text
When comparing the independent predictive power of different data types, clinical data came out on top, not the more technically complex whole slide images or genomic data.
The “digital fingerprint” generated from doctors’ clinical notes alone achieved 98.5% accuracy in 33 cancer classification tasks. This data also performed best when searching for similar patients.
It’s like predicting a car’s performance. You might think you need engine blueprints (genomics) and wind tunnel test data (imaging), but it turns out the most accurate prediction comes from the logbook that records every refueling and repair.
This result suggests that in the age of AI, the value of high-quality, structured clinical data might be underestimated.
Of course, multimodal data still has its place. The researchers found that for more complex problems, like predicting overall survival, integrating all data types produced better results than clinical data alone. This shows that different types of data have different strengths for solving different problems.
Another Surprise: Generalists Beat Specialists
The researchers also tested which AI model was best at interpreting clinical notes. The intuition was that a medical AI trained specifically on medical literature would have an edge.
But the results showed that a general-purpose model (Qwen3) outperformed the specialized medical models.
This might be because clinical records are mostly descriptive natural language, not highly specialized jargon, and general-purpose large language models are very good at handling natural language.
The Honeybee framework is an open-source tool that can integrate multiple types of cancer data. This research also offers two important insights backed by data:
First, clinical data holds enormous potential and its value shouldn’t be overlooked. Second, when choosing AI tools, a general-purpose model might be better than a specialized one for certain tasks.
📜Title: Honeybee: Enabling Scalable Multimodal AI in Oncology Through Foundation Model–Driven Embeddings 📜Paper: https://arxiv.org/abs/2405.07460
3. Boltzina: Making AI Drug Screening Both Fast and Accurate
Virtual screening has long faced a trade-off: it’s hard to get both speed and accuracy.
On one hand, traditional tools like AutoDock Vina are the “fast guns.” They can process millions of molecules quickly, but they have a high false-positive rate and aren’t very precise.
On the other hand, new AI models like Boltz-2 are the “sharpshooters.” They are highly accurate. But their calculations take a long time, making them impractical for screening libraries of millions of molecules.
Researchers have been looking for a tool that offers both speed and accuracy. Boltzina provides a practical solution.
The Fast Gun and the Sharpshooter Team Up
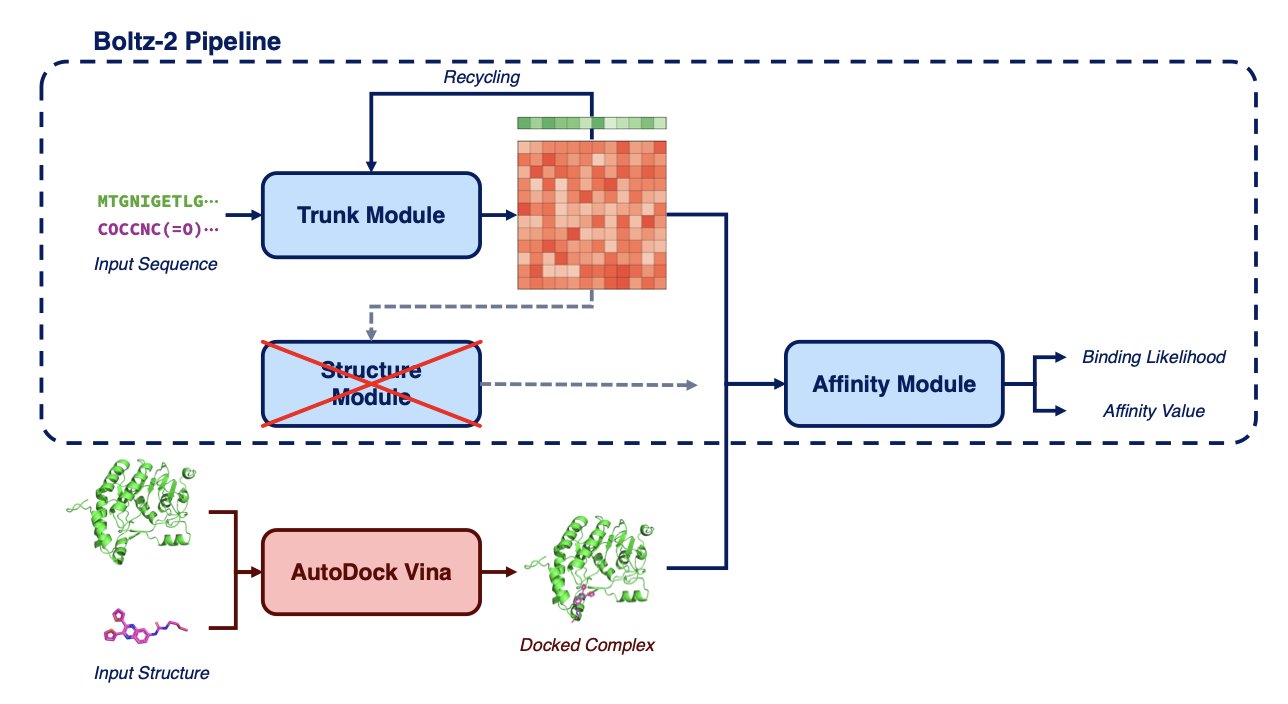
Boltzina’s idea is to re-engineer the workflow, letting the two types of tools work together.
Here’s how it works:
Step one, the “fast gun” Vina takes the stage. It docks a large number of molecules to a protein target, generating all possible binding “poses.” This step is about speed and coverage, not precision.
Step two, the “sharpshooter” Boltz-2 does the final evaluation. Boltzina feeds the poses generated by Vina directly to Boltz-2’s scoring module, which then assesses the reliability of each pose and gives it a score.
Boltzina skips Boltz-2’s most powerful but also most time-consuming “structural co-folding” step, using only its efficient scoring module. It’s like asking an art expert to appraise a batch of existing works without having to sculpt them from scratch.
The Results of the Combined Strategy
This workflow maintains the high screen enrichment rate of Boltz-2 while increasing speed by nearly 12 times. A calculation that used to take a week can now be done in a day.
The researchers also made some optimizations. They found that averaging the Boltzina scores for multiple poses generated by Vina produced more stable and reliable results.
They also proposed a two-stage screening strategy: first, use Boltzina to quickly screen a large compound library down to a few thousand candidates. Then, use the full Boltz-2 to refine the selection from these candidates. For projects with limited resources, this is a reasonable workflow that balances cost and benefit.
Boltzina is an engineering solution. It doesn’t invent a brand-new tool. Instead, it combines two existing tools, each with its own pros and cons, to solve a real problem that researchers face.
It makes the predictive power of models like Boltz-2 practical for the first time on actual screening projects involving hundreds of thousands to millions of compounds.
📜Title: Boltzina: Efficient and Accurate Virtual Screening via Docking-Guided Binding Prediction with Boltz-2 📜Paper: https://arxiv.org/abs/2508.17555v1 💻Code: https://github.com/ohuelab/boltzina